
Abstract
Recent years have seen great advances in our ability to predict crystal structures from first principles. However, previous algorithms have focused on the prediction of bulk crystal structures, where the global minimum is the target. Here, we present a general atomistic approach to simulate in multicomponent systems the structures and free energies of grain boundaries and heterophase interfaces with fixed stoichiometric and non-stoichiometric compositions. The approach combines a new genetic algorithm using empirical interatomic potentials to explore the configurational phase space of boundaries, and thereafter refining structures and free energies with first-principles electronic structure methods. We introduce a structural order parameter to bias the genetic algorithm search away from the global minimum (which would be bulk crystal), while not favouring any particular structure types, unless they lower the energy. We demonstrate the power and efficiency of the algorithm by considering non-stoichiometric grain boundaries in a ternary oxide, SrTiO3.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

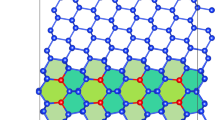
 interface.
interface.

 grain boundary.
grain boundary.
Similar content being viewed by others


Change history
04 March 2010
In the version of this Article originally published, the words ‘an unusual’ in the abstract should have been ‘a new’. This has been corrected in all versions of this Article.
References
Dillon, S. J., Tang, M., Carter, W. C. & Harmer, M. J. Complexion: A new concept for kinetic engineering in materials science. Acta Mater. 55, 6208–6218 (2007).
Sun, E. Y. et al. Microstructural design of silicon nitride with improved fracture toughness: II, effects of yttria and alumina additives. J. Am. Ceram. Soc. 81, 2831–2840 (1998).
Chiang, Y.-M., Silverman, L. A., French, R. H. & Cannon, R. M. Thin glass film between ultrafine conductor particles in thick-film resistors. J. Am. Ceram. Soc. 77, 1143–1152 (1994).
Luo, J., Wang, H. F. & Chiang, Y.-M. Origin of solid-state activated sintering in Bi2O3-doped ZnO. J. Am. Ceram. Soc. 82, 916–920 (1999).
von Alfthan, S., Haynes, P. D., Kaski, K. & Sutton, A. P. Are the structures of twist grain boundaries in silicon ordered at 0 K? Phys. Rev. Lett. 96, 055505 (2006).
Xiang, H. J., Da Silva, J. L. F., Branz, H. M. & Su-Huai, W. Understanding the clean interface between covalent Si and ionic Al2O3 . Phys. Rev. Lett. 103, 116101 (2009).
Deaven, D. M. & Ho, K. M. Molecular geometry optimization with a genetic algorithm. Phys. Rev. Lett. 75, 288–291 (1995).
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 124, 244704 (2006).
Zhang, J., Wang, C. Z. & Ho, K. M. Finding the low-energy structures of Si[001] symmetric tilted grain boundaries with a genetic algorithm. Phys. Rev. B 80, 174102 (2009).
Morris, G. M. et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662 (1998).
Benedek, N. A., Chua, A.-L. S., Elsässer, C., Sutton, A. P. & Finnis, M. W. Interatomic potentials for strontium titanate: An assessment of their transferability and comparison with density functional theory. Phys. Rev. B 78, 064110 (2008).
Zhang, S. B. & Northrup, J. E. Chemical-potential dependence of defect formation energies in GaAs—application to Ga self-diffusion. Phys. Rev. Lett. 67, 2339–2342 (1991).
Finnis, M. W., Lozovoi, A. Y. & Alavi, A. The oxidation of NiAl: What can we learn from ab initio calculations? Annu. Rev. Mater. Res. 35, 167–207 (2005).
Brindle, A. Genetic algorithms for function optimization. Tech. Report TR81-2, (Univ. Alberta, Department of Computer Science, 1981).
Goldberg, D. E. & Deb, K. in Foundations of Genetic Algorithms I (ed. Rawlins, G. J. E.) (Morgan Kaufmann, 1991).
Zhang, Z. L., Sigle, W., Phillipp, F. & Rühle, M. Direct atom-resolved imaging of oxides and their grain boundaries. Science 302, 846–849 (2003).
Steinhardt, P. J., Nelson, D. R. & Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 28, 784–805 (1983).
Batyrev, I. G., Alavi, A. & Finnis, M. W. Equilibrium and adhesion of Nb/sapphire: The effect of oxygen partial pressure. Phys. Rev. B 62, 4698–4706 (2000).
Johnston, K., Castell, M. R., Paxton, A. T. & Finnis, M. W. SrTiO3(2×1) reconstructions: First-principles calculations of surface energy and atomic structure compared with scanning tunnelling microscopy images. Phys. Rev. B 70, 085415 (2004).
Clark, S. J. et al. First principles methods using CASTEP. Z. Kristallogr. 220, 567–570 (2005).
Acknowledgements
This work was supported by the European Commission under contract No. NMP3-CT-2005-013862 (INCEMS). The calculations were carried out using the facilities of the High Performance Computing Service at Imperial College London.
Author information
Authors and Affiliations
Contributions
A.L.-S.C. designed the genetic algorithm (with contributions from all authors), implemented it and carried out the genetic-algorithm simulations. N.A.B. carried out the ab initio thermodynamics calculations and some of the genetic-algorithm simulations. L.C. tested an early version of the genetic algorithm. A.L.-S.C., N.A.B., M.W.F. and A.P.S. wrote and edited the manuscript. A.P.S. conceived the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Information (PDF 188 kb)
Rights and permissions
About this article
Cite this article
Chua, AS., Benedek, N., Chen, L. et al. A genetic algorithm for predicting the structures of interfaces in multicomponent systems. Nature Mater 9, 418–422 (2010). https://doi.org/10.1038/nmat2712
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmat2712
This article is cited by
-
Decorated bacteria-cellulose ultrasonic metasurface
Nature Communications (2023)
-
In-situ scanning transmission electron microscopy study of Al-amorphous SiO2 layer-SiC interface
Journal of Materials Science (2023)
-
Effects of twin thicknesses on incoherent twin-boundary structures in face-centered cubic metals
Science China Materials (2023)
-
An unconstrained approach to systematic structural and energetic screening of materials interfaces
Nature Communications (2022)
-
Direct insight into the structure-property relation of interfaces from constrained crystal structure prediction
Nature Communications (2021)
