
Abstract
Pathogenic microbes have evolved sophisticated molecular strategies to subvert host defenses. Here we show that virulent bacteria interfere directly with Toll-like receptor (TLR) function by secreting inhibitory homologs of the Toll/interleukin-1 receptor (TIR) domain. Genes encoding TIR domain containing–proteins (Tcps) were identified in Escherichia coli CFT073 (TcpC) and Brucella melitensis (TcpB). We found that TcpC is common in the most virulent uropathogenic E. coli strains and promotes bacterial survival and kidney pathology in vivo. In silico analysis predicted significant tertiary structure homology to the TIR domain of human TLR1, and we show that the Tcps impede TLR signaling through the myeloid differentiation factor 88 (MyD88) adaptor protein, owing to direct binding of Tcps to MyD88. Tcps represent a new class of virulence factors that act by inhibiting TLR- and MyD88-specific signaling, thus suppressing innate immunity and increasing virulence.
Similar content being viewed by others


Main
Pathogens increase their virulence and survival time in infected hosts by resisting the antimicrobial host defense. A classical resistance mechanism is encapsulation, which serves to restrict the bactericidal effects of antibodies and complement and promotes invasive infections of the brain and respiratory tract. Another example is antigenic drift, which has been evolved by microbes to keep ahead of the specific immune response and which enables pathogens like Neisseria gonorrhoeae, influenza virus and trypanosomes to survive in immune hosts. Subversion of the fast-acting innate immune response is probably essential for survival during the early stages of infection, but little is known about the possible mechanisms involved. Such mechanisms would allow pathogens to avoid immediate elimination by the host defense and would increase the chances of establishing a critical population size and delivering toxic signals that perturb host tissue functions in the pathogens' favor. This study demonstrates that pathogens directly inhibit TLR signaling by secreting a structural homolog of the signaling domain of the human TLRs.
The TLRs are essential sensors of microbial attack and orchestrate the innate immune defense against many microorganisms1,2. TLR signaling elicits a proinflammatory response characterized by the secretion of cytokines like tumor necrosis factor (TNF), type I and II interferons (IFNs), and chemokines, which in turn control the recruitment of inflammatory cells to infected tissues. It is, therefore, not surprising that TLRs are pivotal for the innate defense against uropathogenic E. coli3, Staphylococcus aureus4, Mycobacterium tuberculosis5, Listeria monocytogenes6 and Chlamydophila pneumoniae7. The TIR domain is crucial for TLR signaling, as first recognized when a TIR point mutation was shown to confer lipopolysaccharide (LPS) unresponsiveness to C3H/HeJ mice8. Other proteins with TIR domains include the TLR adaptor proteins. MyD88 interacts with all TLRs except TLR3, the TIR-associated protein (TIRAP) interacts with TLR2 and TLR4; TIR domain–containing adaptor protein inducing IFN-β (TRIF) allows MyD88-independent TLR3 and TLR4 signaling, and the TRIF-related adaptor molecule (TRAM) participates in TLR4-induced MyD88-independent signaling9. Competition at the level of TIR has been suggested as an efficient mechanism to control TLR signaling10.
Given the central role of the TLRs in host defense, it would not be surprising if microorganisms had evolved mechanisms to interfere with TLR-mediated immune responses. Indeed, two proteins from vaccinia virus, A46R and A52R, have been shown to interfere with IL-1– and TLR4-mediated activation of nuclear factor-κB (NF-κB)11. Specifically, A52R was found to associate with interleukin receptor–associated kinase-2 (IRAK2) and TNF receptor–associated factor-6 (TRAF6)12. The protein was also able to disrupt signaling complexes containing either TRAF6 and TAK1-binding protein-1 or IRAK2 and TIRAP12. In contrast, A46R interacted with TLR4, MyD88, TIRAP, TRIF and TRAM13. Recently, the TIR-like protein A from Salmonella enterica serovar enteritidis was shown to impair TLR- and MyD88-mediated activation of NF-κB and to increase intracellular bacterial accumulation14. These observations suggest that pathogens are capable of directly modifying the TLR-dependent host defenses.
In a screening of bacterial genomes, we detected one gene encoding a TIR-homologous protein of unknown function in B. melitensis and a second one in the uropathogenic E. coli strain CFT073. We report that these Tcps from B. melitensis and E. coli CFT073 impede TLR signaling and the innate host defense, thereby promoting intracellular bacterial survival and tissue pathology in vivo. On the basis of these findings and the molecular epidemiology of clinical isolates from subjects with urinary tract infection (UTI), we propose that bacterial Tcps represent a new class of virulence factors.
Results
Genomic location, structure and function of TcpB and TcpC
Two genes encoding homologs of the human TIR domains were identified in a database search of bacterial genomes (Fig. 1a). A subsequent in silico analysis of their tertiary structure predicted considerable similarity of these homologs to the TIR domain of human TLR1 (Fig. 1b). TcpB was found in B. melitensis, located close to the 5′ attachment site of a putative phe t-RNA island consisting of several operons (Fig. 1a). One gene close to the 3′ attachment site of the island encodes an integrase, but most of the remaining genes have unknown functions. TcpC was found in the uropathogenic E. coli strain CFT073 within a serU island, in an operon containing two genes in the middle of the island and an integrase gene next to the 5′ attachment site. Amino acid sequence analysis of TcpB and TcpC revealed that the TIR domain is located in the C-terminal half of each protein, where both proteins share some sequence homology (Fig. 1c). The N-terminal halves contain no other annotated domains except for a putative transmembrane segment in the case of TcpC. TLR-homologous sequences within the TIR domains of the Tcps include a Box 1 motif, which is present in eukaryotic TIR domains and is essential for signaling (Fig. 1d)15.

(a) Operon structure of CFT073 serU and Brucella phe t-RNA islands. (b) Comparison of the tertiary structure of the TIR domain of human TLR1 to the predicted tertiary structure of TcpC and TcpB (composed using PyMOL Molecular Graphics System, http://www.pymol.org). Att, attachment site. (c) Amino acid sequence comparison of TcpC and TcpB. Identical residues are underlined. (d) Amino acid sequence comparison within Boxes 1–3 of the TIR domains of TLR2, TLR4, MyD88, TIRAP, TcpB and TcpC. Identical residues found in at least two sequences are underlined.
To analyze the function of TcpC during infection, we constructed a tcpC-deletion mutant of CFT073 (tcpC::kan) and the tcpC::kan + pTcpC mutant, which is complemented with a plasmid containing the tcpC operon controlled by its endogenous promoter. The innate response to these strains was studied in the mouse RAW264.7 macrophage cell line and the uroepithelial cell line HCV29. Infection with the tcpC::kan mutant stimulated a much higher TNF (Fig. 2a) or IL-6 response (Fig. 2b) in macrophages or HCV29 cells, respectively, than the wild-type CFT073 strain or the tcpC::kan + pTcpC complemented mutant strain. Analogously, TcpB reduced TNF secretion in RAW264.7 macrophages infected with the mutant tcpC::kan strain or a BL21 E. coli K12 strain complemented with an inducible plasmid encoding tcpB (Fig. 2c,d).

(a,b) TNF secretion by RAW264.7 macrophages (a) or IL-6 secretion by HCV29 uroepithelial cells (b) that were either not infected or infected with E. coli CFT073, the mutant tcpC::kan or the complemented mutant tcpC::kan + pTcpC with the indicated multiplicities of infection (MOIs). Cytokine levels were determined after 5 h of infection. (c) RAW264.7 macrophages were either not infected or infected with the E. coli CFT073 complemented mutant tcpC::kan + pTcpB at the indicated MOIs. TcpB protein expression was induced 2.5 h before tcpC::kan + pTcpB was added to the culture. Uninduced bacteria harboring pTcpB served as control. TNF secretion was determined 5 h upon infection. (d) The experiment in c was repeated using the E. coli strain BL21-CodonPlus-RIL as host for the plasmid pTcpB. Error bars indicate s.d. of three individual cultures. (e,f) Intracellular bacteria in RAW264.7 macrophages (e) or HCV29 cells (f) after 5 h of infection. Extracellular bacteria were killed with gentamicin (50 μg/ml). *P < 0.05, ANOVA on ranks. (g,h) Total number of bacteria per well, determined after 5 h of infection of RAW264.7 macrophages (g) or HCV29 cells (h). All error bars indicate s.d. of three individual experiments.
TcpC was subsequently shown to facilitate the intracellular survival of E. coli CFT073. The wild-type and the complemented mutant tcpC::kan + pTcpC strains had accumulated in higher numbers than the mutant tcpC::kan strain 5 h after infection of RAW264.7 cells and HCV29 cells (Fig. 2e,f), whereas total bacterial numbers were similar (Fig. 2g,h).
Bacterial Tcps affect TLR signaling
TcpC and TcpB were shown to suppress TLR-mediated signaling in NF-κB reporter assays. HEK293 cells were transfected with plasmids encoding TLR4, myeloid differentiation protein-2 (MD-2), an NF-κB reporter construct and TcpC or TcpB. Transfected cells were stimulated with LPS or TNF. TcpC and TcpB inhibited the TLR4-mediated NF-κB response to LPS, but the response to TNF was not affected (Fig. 3a and Supplementary Fig. 1a online). Both proteins also impaired the NF-κB response to the potent TLR2 agonist HSP60 from C. pneumoniae16 in cells transfected with TLR2 (Fig. 3b and Supplementary Fig. 1b). These results show that the Tcps interfere with TLR signaling and suggest that this inhibition may involve the MyD88 adaptor protein.

(a) HEK293 cells were transfected with an NF-κB–luciferase reporter construct (50 ng/ml) and TLR4- (2.5 ng/ml), MD2- (2.5 ng/ml) or TcpC-encoding plasmids as indicated. Twenty-four hours later, cells were stimulated with LPS (100 ng/ml) or TNF (10 ng/ml) for 24 h. (b) The experiment was performed as in a except that cells were transfected with TLR2 (4 ng/ml) or TcpC (8 ng/ml) and stimulated with HSP60 (20 μg/ml). Unstimulated cells in a and b served as controls. Luciferase activity was normalized to cells transfected with empty vector (e.v.). Error bars in a and b depict s.d. from three individual cultures. (c) Cell lysates were prepared from HEK293 cells that were transfected or not transfected with myc-tagged MyD88 as indicated. Pull-down assays of the cellular lysates were performed with TIR-TcpC agarose (+) or empty agarose (−). Bound proteins were washed three times with 0.03 or 0.5 M NaCl, eluted and analyzed by western blotting using a MyD88-specific antiserum or a myc-specific monoclonal antibody (anti-myc). An antiserum specific for GFP was used as isotype control. (d) RAW264.7 cells were stimulated for the indicated time with the tcpC::kan mutant E. coli. Pull-down assays were performed with concentrated cell lysates and analyzed by western blotting using a MyD88 antiserum. Expression of MyD88 was analyzed in cellular lysates. β-actin expression served as loading control. (e) The experiment was performed as described in c, but pull-down assays were performed in the absence or presence of TcpB. (f) Wild-type or MyD88-deficient BMMs were uninfected (mock) or infected with the mutant tcpC::kan or CFT073 E. coli at the indicated MOIs. TNF secretion was analyzed 5 h after infection. Error bars represent s.d. from three individual cultures. (g,h) Wild-type (g) and MyD88-deficient (h) BMMs were infected with tcpC::kan, tcpC::kan + pTcpC or CFT073 E. coli at the indicated MOIs. Intracellular bacteria were quantified 5 h later. Error bars indicate s.d. of three individual experiments. *P < 0.05, ANOVA on ranks.
To test this hypothesis, we overexpressed MyD88 in HEK293 cells and quantified the influence of TcpB on NF-κB activation. TcpB efficiently blocked MyD88-induced activation of NF-κB (Supplementary Fig. 1c). Furthermore, TcpB inhibited NF-κB activation induced by coexpression of IRAK1 and IRAK4 or MyD88, IRAK1 and IRAK4 (Supplementary Fig. 1c). However, TcpB did not impair TLR3-TRIF–mediated activation of the IFN-β promoter by the TLR3 ligand poly(I:C), as shown in HEK293 cells transfected with an IFN-β promoter reporter construct, TLR3 and TcpB (Supplementary Fig. 1d).
TcpC was subsequently shown to bind to MyD88 in pull-down assays using the purified TIR domain of TcpC (TIR-TcpC). TIR-TcpC interacted with transfected and endogenous MyD88 of HEK293 cells (Fig. 3c). Furthermore, the endogenous MyD88 of RAW264.7 cells bound to TIR-TcpC when the cells were prestimulated with the tcpC::kan mutant bacteria for at least 15 min (Fig. 3d). Prestimulation of RAW264.7 cells was required to increase endogenous MyD88 expression (Fig. 3d). TIR-TcpC did not interact with other components of the TLR signaling cascade, as shown by pull-down assays with IRAK1, IRAK4 (Supplementary Fig. 2a online), TRIF and the intracellular domain of TLR2 (Supplementary Fig. 2b). TcpB shared the ability of TIR-TcpC to bind endogenous and transfected MyD88 (Fig. 3e).
Subsequent experiments using wild-type or MyD88-deficient bone marrow–derived macrophages (BMMs) infected with E. coli CFT073 or the tcpC::kan mutant addressed whether MyD88 is required for the function of TcpC. Although TcpC suppressed TNF secretion by MyD88-positive BMMs, it failed to do so in MyD88-deficient BMMs (Fig. 3f). Furthermore, the difference in intracellular accumulation between the tcpC::kan mutant E. coli and E. coli CFT073 or the complemented tcpC::kan + pTcpC mutant E. coli was seen in wild-type but not MyD88-deficient BMMs (Fig. 3g,h). Taken together, these results show that TcpC interacts and interferes with the MyD88-dependent effector functions of innate immunity.
TcpC is involved in the pathogenesis of pyelonephritis
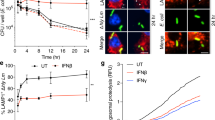
We observed a direct effect of TcpC on the pathogenesis of acute pyelonephritis after comparing the wild-type or mutant tcpC::kan strains in the mouse UTI model, in which we saw a difference between the two strains in bacterial burden and tissue damage. The wild-type multiplied to very high numbers, as shown by cultures of urine and kidney homogenates (Fig. 4a,b). The complemented tcpC::kan + pTcpC mutant strain, in contrast, reached bacterial numbers in the kidneys similar to those of the CFT073 strain after 24 h (Fig. 4c). Furthermore, kidney abscesses were detected in mice after infection with CFT073, but not with the tcpC::kan mutant (Fig. 4d–f). By staining for P-fimbriae, a marker of uropathogenic E. coli, we detected bacteria in the centers of the abscesses, which contained numerous neutrophils (Fig. 4g).

(a,b) Bacterial burden in urine (a) and kidneys (b) after infection of C57BL/6 mice (8–10 mice per time point) with CFT073 or tcpC::kan mutant E. coli. CFU, colony-forming units. (c) Bacterial numbers in the kidneys,24 h after infection with CFT073, tcpC::kan or tcpC::kan + pTcpC E. coli (*P < 0.05, for CFT073 and tcpC::kan + pTcpC versus tcpC::kan, Fisher's exact test). (d,e) Macroscopic abscesses (dotted line) in cross-sections of mouse kidneys after infection with CFT073 (d) or tcpC::kan mutant (e) E. coli. Scale bar, 2 mm. (f,g) Histology of kidney tissue stained with htx-eosin after paraformaldehyde fixation (f) or with specific antibodies to neutrophil granulocytes (green, Rb68C5) and the PapG adhesin (red, antiserum to a synthetic PapG peptide). Scale bar, 200 μm. (g). The abscess is indicated by the white dotted line. Scale bar, 200 μm. (h) TcpC is common in the most virulent uropathogenic E. coli strains from children with acute pyelonephritis (n = 101), is less common in acute cystitis (n = 58) and asymptomatic bacteriuria (ABU, n = 77), and is rare in fecal E. coli strains (FN, n = 39). TcpC was detected by PCR. *P = 0.016 and P = 0.001, Fisher's exact test.
We next examined the molecular epidemiology of tcpC in human UTI. E. coli strains were isolated from the urine of individuals with severe kidney infections (acute pyelonephritis), bladder infections (acute cystitis) or asymptomatic bacterial carriage (asymptomatic bacteriuria). Isolates from the fecal flora of individuals without UTI were used as commensal E. coli controls. TcpC-homologous sequences were present in about 40% of acute pyelonephritis isolates but were less common in cystitis (21%), asymptomatic bacteriuria (16%) or commensal (8%) E. coli strains (Fig. 4h). The results suggest that the tcpC sequences enhance virulence, as indicated by their association with the clinical severity of UTI.
TcpC is secreted by E. coli CFT073
To determine whether TcpC is secreted, we generated a polyclonal antiserum to TcpC, which detected TcpC in CFT073 strain lysates, but not in the lysates of the tcpC::kan mutant strain (Fig. 5a). TcpC production was enhanced by lowering the pH of the culture medium or by co-incubation of E. coli CFT073 with RAW264.7 cells (Fig. 5a). We also detected TcpC in the culture supernatant of CFT073-infected HCV29 and RAW264.7 cells (Supplementary Fig. 3a online) but did not detect DnaK, an intracellular chaperone protein (Supplementary Fig. 3b), making it unlikely that TcpC was released during bacterial lysis. Using a transwell system, we found that E. coli CFT073 physically separated from BMMs suppressed TNF induction when compared to the tcpC::kan mutant (Fig. 5b). We verified that bacteria did not cross the membrane of the transwell by culturing the supernatant of the BMMs. In addition, TcpC was present in equal amounts inside BMMs compared to a non-separated coculture as determined by western blotting (Fig. 5c). Confocal microscopy using a tetracysteine (FlAsH)-tagged variant of TcpC (details are described in the Supplementary Methods online) confirmed this finding and clearly showed that TcpC was detectable within host cells (Fig. 5d). Because certain bacterial virulence factors are known to interact with lipid rafts rich in cholesterol, we used the cholesterol-extracting agent methyl-β-cyclodextrin (MβCD)17 to block the cellular uptake of TcpC. The compound impaired the uptake of recombinant TIR-TcpC, and extracellular TcpC was detectable only in the presence of MβCD (Fig. 5e). Taken together, the results show that E. coli CFT073 secretes TcpC and that the secreted and recombinant TcpC protein is taken up by host cells.

(a) Spontaneous and induced TcpC expression by CFT073 upon culture in medium, medium at pH 5 or upon coculture with RAW264.7 cells for 5 h. The mutant tcpC::kan E. coli was used as negative control. (b) BMMs were cocultured in transwell plates with mutant tcpC::kan or CFT073 E. coli at the indicated MOIs. BMMs cultured in the absence of bacteria served as controls. TNF secretion was analyzed 5 h after infection. Error bars represent s.d. from three individual cultures. (c) Detection of intracellular TcpC by western blotting after coculture of RAW264.7 cells with E. coli CFT073 (MOI 1) or with the mutant tcpC::kan strain (MOI 1) in normal or transwell plates. Uninfected cells (mock) served as negative controls. BMMs were trypsinized after 5 h of culture, washed and lysed with RIPA buffer. (d) RAW264.7 cells were cultured in transwell plates with the mutant tcpC::kan strain complemented (left) or not complemented (right) with pFlAsH-TcpC at an MOI of 5. Immediately before infection, FlAsH-EDT2 reagent was added to the culture, and TcpC accumulation in macrophages was monitored by confocal microscopy 90 min after infection. (e) BMMs were incubated with different doses of TIR-TcpC in the presence or absence of MβCD (10 mM) as indicated in the graph. After 2.5 h of culture, intra- and extracellular TIR-TcpC was detected by western blotting of the cell lysate and concentrated culture supernatant, respectively.
The data above suggest that secreted TcpC is sufficient to impair the cytokine response of innate immune cells. To confirm this, we stimulated RAW264.7 cells with a variety of TLR-ligands in a dose-dependent manner and observed that recombinant TIR-TcpC inhibited TNF release by the cells (Fig. 6a). There was efficient inhibition of the MyD88-dependent TNF response to LPS, but not of the TNF response to poly(I:C), which involves TRIF rather than MyD88 (Fig. 6a). Again, we found recombinant TIR-TcpC inside the cells (Supplementary Fig. 4 online). Recombinant EGFP purified by the same method as TIR-TcpC did not interfere with endotoxin-induced TNF-secretion (Fig. 6b). These results confirmed the previous observation that TcpB impaired TLR2- and TLR4-induced activation of NF-κB but not TLR3-driven activation of the IFN-β promoter (Supplementary Fig. 1). MβCD reversed the inhibition of LPS-induced signaling by TIR-TcpC, indicating that uptake of TIR-TcpC was required for its function (Fig. 6c).

(a) RAW264.7 macrophages were stimulated with Pam3Cys (1 μg/ml), poly(I:C) (2.5 μg/ml), ultrapure LPS from E. coli K12 (100 ng/ml), flagellin from Salmonella typhimurium (1 μg/ml) and CpG 1826 (2 μM) in the presence of titrated amounts of the purified TIR domain of TcpC (TIR-TcpC) as indicated. TNF secretion was analyzed 3 h after stimulation. (b) To control for nonspecific suppressive effects, cells were stimulated with ultrapure LPS from E.coli K12 (100 ng/ml) in the presence of titrated amounts of recombinant EGFP expressed and purified by the same method as used for TIR-TcpC. TNF was determined after 5 h of culture. Error bars depict s.d. from three individual cultures. (c) BMMs were stimulated with ultrapure LPS from E. coli K12 (100 ng/ml) in the presence or absence of MβCD (10 mM) and different doses of TIR-TcpC as indicated. TNF abundance was determined after 2.5 h of culture. (d) RAW264.7 cells were infected with E. coli CFT073 or the mutant tcpC::kan strain for 5 h at the indicated MOIs in the absence or presence of the efflux pump inhibitor PAβN (52 μM). Error bars indicate s.d. of three individual cultures. (e) RAW264.7 cells were infected with E. coli CFT073 (MOI 1) or the mutant tcpC::kan strain (MOI 1) or were not infected (mock) in the presence or absence of PAβN (52 μM) as indicated. TcpC was detected by western blotting after 5 h of culture.
Because TcpC is secreted, we tested whether the efflux pump inhibitor phenylalanine-arginine-β-naphtylamide (PAβN)18 could neutralize the effect of TcpC and therefore serve as a potential therapeutic drug. PAβN blocked the ability of E. coli CFT073 to inhibit TNF production by RAW264.7 cells but did not influence the TNF secretion induced by the tcp::kan mutant (Fig. 6d). Furthermore, the compound impaired the secretion of TcpC by the CFT073 strain (Fig. 6e). Thus, PAβN could be envisioned as an additional treatment strategy to accompany antibiotics in severe UTI.
Discussion
This study provides strong evidence that human pathogens produce TLR homologs to inhibit TLR signaling and promote survival. We identified two bacterial homologs of the TIR signaling domain in uropathogenic E. coli and B. melitensis. TcpC was shown to be associated with human disease by molecular epidemiological studies and was also shown to increase bacterial burden and tissue damage in the mouse UTI model. Furthermore, TcpC from E. coli CFT073 promoted the intracellular accumulation of bacteria. In vitro, TcpC and TcpB impaired TLR signaling and the secretion of proinflammatory cytokines during infection of host cells. Both Tcps interacted with MyD88, resulting in the inhibition of MyD88-induced activation. TcpC thus acts as an independent virulence factor that is secreted and taken up by host cells, where it impairs cytokine production. These findings also suggest a new treatment modality for severe UTI, as the efflux pump inhibitor PAβN reduces the secretion of TcpC and thus normalizes the inflammatory response to E. coli CFT073. The results identify the TIR-homologous proteins as a noteworthy example of molecular mimicry and add the Tcps to a very limited number of molecules that directly subvert TLR signaling and impair the activation of the innate host defense.
TIR-homologous genes are found in several bacterial species but are not ubiquitously distributed throughout the bacterial kingdom, and their potential as virulence factors has not been defined. We provide evidence that TcpC is clinically relevant as a virulence factor in the subset of E. coli strains that cause severe kidney infections. TcpC was prevalent in isolates from children with acute pyelonephritis, but rare in environmental E. coli strains from the fecal flora of healthy children and in E. coli strains causing the less severe forms of UTI. This association is highly relevant, as TLR4 controls the antibacterial defenses of the urinary tract19,20.
Our database search revealed that aside from E. coli CFT073 and B. melitensis, other human pathogens also harbor related genes, including B. suis and B. abortus and the S. aureus strain MSSA476. Brucellosis is a zoonotic disease characterized by undulating fever, enlargement of lymph nodes and hepatosplenomegaly. Brucella species are facultative intracellular bacteria that reside intracellularly in early endosomes, prevent the fusion of phagosomes with lysosomes and replicate within the endoplasmic reticulum21. The molecular mechanisms of this intracellular persistence are only partially understood, but the virB operon, which encodes a type IV secretion system, is necessary for intracellular survival, as virB mutants are defective in this respect22,23. Furthermore, Brucella species possess an atypical endotoxin, characterized by a diaminoglucose backbone and by longer acyl groups than E. coli endotoxin24, that is weakly stimulatory and prevents complement-mediated lysis of the bacteria. The endotoxin seems to be important for lipid raft–mediated uptake of the bacteria by host cells and for the early phase of intracellular transport of Brucella25,26. On the basis of the present study, it seems that TcpB may help Brucella avoid recognition by innate immune cells by further decreasing the TLR-dependent response to infection.
We also found that TcpC is secreted, and the purified TIR domain of the protein is sufficient to impede TNF secretion by macrophages stimulated with different TLR ligands. At present, the molecular mechanism of TcpC secretion is not known, but the involvement of a type III secretion system can be excluded because the genome of CFT073 does not contain the necessary genes27. Also, TcpC does not contain the classical sec leader sequence required for type II secretion; however, a functional type I secretion system has been described for E. coli CFT073 (ref. 27). These results suggest that uropathogenic E. coli influences TLR signaling from the exterior of the cell. Fimbriae-mediated adherence was not needed for this to occur, but it is probable that the fimbriae increase the efficiency of TcpC by bringing the bacteria close to the host cell surface. Secreted TcpC, as well as the purified recombinant protein, accumulated intracellularly to inhibit host TLR signaling. On the basis of these results, we used the efflux pump inhibitor PAβN, which is known to reverse multidrug resistance against antibiotics in E. coli and other Enterobacteriaceae28,29, as a potential therapeutic compound. The experiments show that the virulence of E. coli CFT073, as indicated by its ability to impair TNF secretion by macrophages, can be efficiently reduced with this drug.
We have found that bacterial Tcps suppress MyD88-dependent, but not TRIF-dependent, signaling pathways. TcpC and TcpB both impaired TLR2- and TLR4-mediated activation of NF-κB and interacted with MyD88. TcpB did not affect TLR3-mediated activation of the IFN-β promoter, which is consistent with the MyD88 independence of this response, and recombinant TIR-TcpC did not modulate TLR3-mediated TNF release or interact with TRIF but did affect the TNF response to TLR2, TLR4, TLR5 and TLR9 ligands. Moreover, infection of BMMs with E. coli CFT073 attenuated cytokine secretion and increased the accumulation of intracellular bacteria in wild-type host cells but not in MyD88-deficient cells. In contrast, the TIR-containing vaccinia virus protein A46R interacts with TLR4 and all four adaptor molecules of the TLR signaling cascade12,13. The results suggest that bacteria are able to modify the host defenses from a distance, before contact with the mucosa. Through this mechanism, they are likely to gain enough time to further perturb the host defense in favor of their own survival.
Methods
E. coli strains.
We obtained E. coli isolates from children with their first defined episode of acute pyelonephritis or acute cystitis, isolates from asymptomatic carriers after screening for bacteriuria in female children, and fecal isolates from healthy children without a history of UTI. We maintained the strains as deep agar stabs and subcultured them on TSA plates for DNA extraction. The study was approved by the Goteborg Medical Ethics committee. We purchased the E. coli strain BL21-CodonPlus-RIL from Stratagene. TcpC was detected by PCR using the primers tcpC for: 5′-GGCAACAATATGTATAATATCCT-3′ and tcpC rev: 5′-GCCCAGTCTATTTCTGCTAAAGA-3′.
Mouse urinary tract infection model.
We used female C57BL/6 mice at an age of 8–16 weeks and injected E. coli into the urinary tract as described30. In brief, we installed 0.1 ml of the bacterial suspension (1 × 109 colony-forming units/ml) into the bladder of anesthetized mice through a soft polyethylene catheter (0.61 mm outer diameter; Clay Adams). We obtained urine samples daily and killed the mice after 7 d. We obtained bacterial tissue counts after homogenization and plating. We fixed tissues in paraformaldehyde and stained them with htx-eosin for histology. For immunohistochemistry, we used antibodies to neutrophils (Rb6-8C5) with DAPI counterstaining to visualize cell nuclei. We detected P-fimbriated E. coli with an antiserum to a peptide within the PapG adhesin. The experiments were performed with the permission of the Animal Experimental ethics committee at the Lund District Court, Sweden (numbers M166-04 and M87-079).
Cell lines and bone marrow–derived macrophages.
We obtained RAW 264.7 and HEK293 cells from American Culture Type Collection. S. Schubert provided the human uroepithelial cell line HCV29 (ref. 31). The preparation of mouse bone marrow macrophages is described in the Supplementary Methods.
Antisera, monoclonal antibodies and enzyme-linked immunosorbent assays.
We generated an antiserum against TcpC by immunizing rabbits with the peptides EQTLEVGDSLRRNIDL and FLNKKWTQYELDSLIC (Eurogentec). To quantify TNF and IL-6 in culture supernatants, we applied ELISA Duo sets (R&D Systems). We obtained the antibody to DnaK from Stressgen, antibody to myc label from Invitrogen and MyD88 (N19)- and GFP-specific (I16) antibodies from Santa Cruz Biotechnology. Antibody to mouse β-actin and flag-specific antibody came from Sigma, Strep-Tactin–horseradish peroxidase conjugate from IBA, and horseradish peroxidase–labeled antibodies specific to mouse or rabbit immunoglobulins from Dianova.
Cloning, production and purification of TcpB and TcpC and construction of the CFT073 tcpC::kan mutant E. coli strain.
We amplified tcpB and tcpC by PCR from genomic DNA of B. melitensis or the E. coli strain CFT073, respectively, and cloned them into different eukaryotic and prokaryotic expression plasmids (described in the Supplementary Methods). The creation of tetracysteine-tagged TcpC fusion proteins is described in the Supplementary Methods.
We constructed the E. coli CFT073 tcpC::kan mutant strain using the lambda Red recombinase system32 as described in the Supplementary Methods. Generation of the plasmid pTcpC to complement the mutant is also described in the Supplementary Methods.
Confocal microscopy.
We analyzed the uptake of TcpC into RAW264.7 cells by confocal microscopy. Details are described in the Supplementary Methods.
Induction of TcpC expression in E. coli CFT073.
To induce expression of TcpC, we grew the E. coli CFT073 strain in LB medium at 30 °C to an optical density at 600 nm of 0.5. We transferred bacteria (1 × 107) to a six-well plate and cultured them in the presence of RPMI or RPMI acidified to pH 5 or cocultured them with RAW264.7 cells (2 × 106 cells/well) at 37 °C. After 5 h, we harvested the supernatants, centrifuged them and concentrated them 50-fold using a Nanosep 3K Omega (PALL); then we separated the proteins by SDS-PAGE and analyzed them by western blotting.
Inhibition of TIR-TcpC uptake by methyl-β-cyclodextrin.
We incubated BMMs with MβCD (10 mM, Sigma) for 30 min before adding different amounts of recombinant TIR-TcpC. We treated the cells with trypsin (15 min, 50 μg/ml), washed them three times with PBS and subsequently lysed them with RIPA buffer (Na2HPO4 (pH 7.2–7.4, 10 mM), NaF (50 mM), NaCl (150 mM), EDTA (2 mM), SDS (1% (wt/vol)), sodiumdeoxycholate (1% (wt/vol)), NP-40 (1% (vol/vol)), protease inhibitors (Roche), NaVO3 (0.2 mM)). We detected intra- and extracellular TIR-TcpC by western blotting.
Infection and stimulation assays.
We seeded BMMs, RAW264.7 cells and HCV29 cells in six-well plates at concentrations between 1.5 × 106 and 2.0 × 106 cells per well in DMEM (primary cells) or RPMI-1640 supplemented with 5% FCS, 1% penicillin-streptomycin and 0.1% 2-mercaptoethanol. Immediately before each assay, we washed the cells and added new antibiotic-free medium (1–5% FCS). We then either infected the cells with varying amounts of bacteria (usually for 5 h) or stimulated them with TLR ligands in the absence or presence of the purified recombinant TIR domain of TcpC (TIR-TcpC) for 2 h. We used culture supernatants for quantification of TNF by ELISA or concentrated them 20-fold to detect TIR-TcpC by western blotting. In infection experiments, we killed extracellular bacteria after collecting the supernatant by adding 50 μg/ml gentamicin in PBS. In TIR-TcpC coculture experiments, we washed the cells with PBS and collected them after trypsinization (250 μg/well, 10 min) to avoid the possibility that the TIR-TcpC was stuck to the cell surface. After two additional wash steps in PBS, we lysed the cells with 1 ml NP-40 (1% Igepal) or RIPA buffer supplemented with protease inhibitors (Roche).
For analysis of TcpC secretion, we used transwell plates with 0.4-μm pore size filters (Corning) to separate bacteria from the cultured cells.
In some experiments, we added the efflux pump inhibitor PAβN (Sigma) at a concentration of 52 μM during infection.
Pull-down experiments.
We used purified TcpC-TIR, TcpB or EGFP each carrying a C-terminal Strep-tag II as as 'bait' protein using a biotinylated protein-protein interaction kit (Pierce). We bound 100 μg of purified TIR-TcpC, TcpB or EGFP to the agarose-coupled streptavidin for 1 h at 4 °C. After two blocking steps, we added lysates from HEK293 cells either transfected or non-transfected with MyD88-myc, IRAK1-flag, IRAK4-flag, TRIF-flag, or ICD-TLR2-flag plasmids to the column (1 h at 4 °C followed by 30 min at 22 °C). To prepare the lysates, we solubilized the cells by intensive homogenization in PBS buffer followed by a final sonification step. In some experiments, we treated HEK293 lysates with 0.125% (wt/vol) N-octylglucoside. We solubilized lysates of RAW264.7 cells using NP-40 buffer supplemented with 0.125% (wt/vol) N-octylglucoside followed by dialysis against PBS. We performed three wash steps using an acetate buffer with 0.025 M or 0.5 M NaCl. We eluted the bound 'prey' proteins in three consecutive steps using an elution buffer of pH 2.8 and subsequently collected them in neutralization buffer (2 M TrisCl pH 8.0) before analyzing them by SDS-PAGE or western blotting.
Luciferase reporter assays.
Details on reporter assays are described in the Supplementary Methods.
SDS-PAGE and western blotting.
We used a rabbit polyclonal antiserum (diluted 1:1,000) to detect TcpC in lysates from CFT073 infections that had been resolved on 15% SDS gels. Antibodies specific to rabbit IgG coupled with peroxidase (Jackson Immunoresearch) served as secondary antibody (diluted 1:7,000). We washed blots two times with TBS-T and once with TBS and visualized them with enhanced chemiluminescent reagent (NEN Life Science Products).
Statistical analyses.
Details on the software used and the statistical analysis are provided in the Supplementary Methods.
Note: Supplementary information is available on the Nature Medicine website.
References
Iwasaki, A. & Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 5, 987–995 (2004).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Svanborg, C. et al. Uropathogenic Escherichia coli as a model of host-parasite interaction. Curr. Opin. Microbiol. 9, 33–39 (2006).
Takeuchi, O., Hoshino, K. & Akira, S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to staphylococcus aureus infection. J. Immunol. 165, 5392–5396 (2000).
Abel, B. et al. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J. Immunol. 169, 3155–3162 (2002).
Torres, D. et al. Toll-like receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect. Immun. 72, 2131–2139 (2004).
Rodriguez, N. et al. Differential involvement of TLR2 and TLR4 in host survival during pulmonary infection with Chlamydia pneumoniae. Eur. J. Immunol. 36, 1145–1155 (2006).
Poltorak, A. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 (1998).
Kawai, T. & Akira, S. TLR signaling. Cell Death Differ. 13, 816–825 (2006).
Burns, K. et al. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J. Exp. Med. 197, 263–268 (2003).
Bowie, A. et al. A46R and A52R from vaccinia virus are antagonists of host IL-1 and Toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 97, 10162–10167 (2000).
Harte, M.T. et al. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 197, 343–351 (2003).
Stack, J. et al. Vaccinia virus protein A46R targets multiple Toll-like–interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 201, 1007–1018 (2005).
Newman, R.M., Salunkhe, P., Godzik, A. & Reed, J.C. Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian Toll/interleukin-1 receptor family proteins. Infect. Immun. 74, 594–601 (2006).
Radons, J. et al. The interleukin 1 (IL-1) receptor accessory protein Toll/IL-1 receptor domain: analysis of putative interaction sites in vitro mutagenesis and molecular modeling. J. Biol. Chem. 278, 49145–49153 (2003).
Costa, C.P. et al. Role of chlamydial heat shock protein 60 in the stimulation of innate immune cells by Chlamydia pneumoniae. Eur. J. Immunol. 32, 2460–2470 (2002).
Lafont, F. & van der Goot, F.G. Bacterial invasion via lipid rafts. Cell. Microbiol. 7, 613–620 (2005).
Pannek, S. et al. Multidrug efflux inhibition in Acinetobacter baumannii: comparison between 1-(1-naphthylmethyl)-piperazine and phenyl-arginine-β-naphthylamide. J. Antimicrob. Chemother. 57, 970–974 (2006).
Frendeus, B. et al. Escherichia coli P fimbriae utilize the Toll-like receptor 4 pathway for cell activation. Mol. Microbiol. 40, 37–51 (2001).
Samuelsson, P., Hang, L., Wullt, B., Irjala, H. & Svanborg, C. Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect. Immun. 72, 3179–3186 (2004).
Gorvel, J.P. & Moreno, E. Brucella intracellular life: from invasion to intracellular replication. Vet. Microbiol. 90, 281–297 (2002).
Delrue, R.M. et al. Identification of Brucella spp. genes involved in intracellular trafficking. Cell. Microbiol. 3, 487–497 (2001).
Comerci, D.J., Martinez-Lorenzo, M.J., Sieira, R., Gorvel, J.P. & Ugalde, R.A. Essential role of the VirB machinery in the maturation of the Brucella abortus–containing vacuole. Cell. Microbiol. 3, 159–168 (2001).
Lapaque, N., Moriyon, I., Moreno, E. & Gorvel, J.P. Brucella lipopolysaccharide acts as a virulence factor. Curr. Opin. Microbiol. 8, 60–66 (2005).
Sola-Landa, A. et al. A two-component regulatory system playing a critical role in plant pathogens and endosymbionts is present in Brucella abortus and controls cell invasion and virulence. Mol. Microbiol. 29, 125–138 (1998).
Detilleux, P.G., Deyoe, B.L. & Cheville, N.F. Penetration and intracellular growth of Brucella abortus in nonphagocytic cells in vitro. Infect. Immun. 58, 2320–2328 (1990).
Welch, R.A. et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 99, 17020–17024 (2002).
Kern, W.V. et al. Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Escherichia coli. J. Antimicrob. Chemother. 57, 339–343 (2006).
Schumacher, A. et al. Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Enterobacteriaceae other than Escherichia coli. J. Antimicrob. Chemother. 57, 344–348 (2006).
Hagberg, L. et al. Contribution of adhesion to bacterial persistence in the mouse urinary tract. Infect. Immun. 40, 265–272 (1983).
Bean, M.A., Pees, H., Fogh, J.E., Grabstald, H. & Oettgen, H.F. Cytotoxicity of lymphocytes from patients with cancer of the urinary bladder: detection by a 3- H-proline microcytotoxicity test. Int. J. Cancer 14, 186–197 (1974).
Datsenko, K.A. & Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645 (2000).
Acknowledgements
S. Schubert (Pettenkofer Institut, München, Germany) provided the human uroepithelial cell line HCV29. This work was supported by SFB576, project B10, Deutsche Forschungsgemeinschaft and the Swedish Medical Research Council (grant numbers 07934, 14577, 14578); The Royal Physiographic Society; The Medical Faculty, Lund University; Network of Excellence Europathogenomics; and The Österlund, Lundberg and Wallenberg Foundations. C.S. was a recipient of the Bristol-Myers Squibb unrestricted grant. We thank G. Häcker for critical reading of the manuscript and C. Kirschning (Institut für Medizinische Mikrobiologie, Immunologie und Hygiene, München, Germany), K. Rückdeschel (Institut für Medizinische Mikrobiologie, Virologie und Hygiene, Hamburg, Germany), F. Schmitz (Institut für Medizinische Mikrobiologie, Immunologie und Hygiene, München, Germany) and S. Akira (Research Institute for Microbial Diseases, Osaka, Japan) for the donation of plasmids. We are grateful to S. Bierl and T. Ertl for construction of expression plasmids and experimental assistance.
Author information
Authors and Affiliations
Contributions
C.C. performed pull-down assays, western blotting, cell culture assays and protein purification. A.W. and S.S. generated the E. coli deletion mutant tcpC::kan. M.Y. performed in vivo experiments. S.D. performed luciferase reporter assays and assisted with confocal microscopy. H.F. and D.S. performed PCR analysis of E. coli strains obtained from human beings. N.W. and N.R. assisted in cell culture assays and provided gene-deficient mice. H.W. participated in writing the manuscript. C.S. supervised in vivo experiments and participated in writing the manuscript. T.M. analyzed the data, designed the whole project and wrote the manuscript.
Corresponding author
Supplementary information
Supplementary Text and Figures
Supplementary Figs. 1–4 and Supplementary Methods (PDF 9201 kb)
Rights and permissions
About this article
Cite this article
Cirl, C., Wieser, A., Yadav, M. et al. Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain–containing proteins. Nat Med 14, 399–406 (2008). https://doi.org/10.1038/nm1734
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1734
This article is cited by
-
Uropathogen and host responses in pyelonephritis
Nature Reviews Nephrology (2023)
-
Immunomodulation therapy offers new molecular strategies to treat UTI
Nature Reviews Urology (2022)
-
The prevalence of the iutA and ibeA genes in Escherichia coli isolates from severe and non-severe patients with bacteremic acute biliary tract infection is significantly different
Gut Pathogens (2021)
-
TcpC inhibits neutrophil extracellular trap formation by enhancing ubiquitination mediated degradation of peptidylarginine deiminase 4
Nature Communications (2021)
-
Molecular determinants of disease severity in urinary tract infection
Nature Reviews Urology (2021)
