
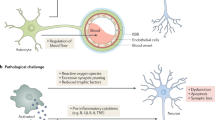
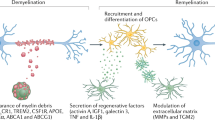
Abstract
There has been an explosion of new findings recently giving us insights into the involvement of microglia in central nervous system (CNS) disorders. A host of new molecular tools and mouse models of disease are increasingly implicating this enigmatic type of nervous system cell as a key player in conditions ranging from neurodevelopmental disorders such as autism to neurodegenerative disorders such as Alzheimer's disease and chronic pain. Contemporaneously, diverse roles are emerging for microglia in the healthy brain, from sculpting developing neuronal circuits to guiding learning-associated plasticity. Understanding the physiological functions of these cells is crucial to determining their roles in disease. Here we focus on recent developments in our rapidly expanding understanding of the function, as well as the dysfunction, of microglia in disorders of the CNS.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Debbie Maizels/Springer Nature

Debbie Maizels/Springer Nature

Debbie Maizels/Springer Nature
Similar content being viewed by others

References
Kierdorf, K. et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 16, 273–280 (2013).
Ginhoux, F. et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010).
Gomez Perdiguero, E. et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551 (2015).
Sheng, J., Ruedl, C. & Karjalainen, K. Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 43, 382–393 (2015).
Hoeffel, G. et al. C-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678 (2015).
Hagemeyer, N. et al. Transcriptome-based profiling of yolk sac-derived macrophages reveals a role for Irf8 in macrophage maturation. EMBO J. 35, 1730–1744 (2016).
Gosselin, D. et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 (2014).
Minten, C., Terry, R., Deffrasnes, C., King, N.J. & Campbell, I.L. IFN regulatory factor 8 is a key constitutive determinant of the morphological and molecular properties of microglia in the CNS. PLoS One 7, e49851 (2012).
Goldmann, T. et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805 (2016).
Matcovitch-Natan, O. et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 353, aad8670 (2016).
Grabert, K. et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516 (2016).
Pannell, M. et al. The subpopulation of microglia expressing functional muscarinic acetylcholine receptors expands in stroke and Alzheimer's disease. Brain Struct. Funct. 221, 1157–1172 (2016).
Wlodarczyk, A. et al. Pathologic and protective roles for microglial subsets and bone marrow- and blood-derived myeloid cells in central nervous system inflammation. Front. Immunol. 6, 463 (2015).
Hashimoto, D. et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804 (2013).
Bruttger, J. et al. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 43, 92–106 (2015).
Lawson, L.J., Perry, V.H. & Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48, 405–415 (1992).
Erblich, B., Zhu, L., Etgen, A.M., Dobrenis, K. & Pollard, J.W. Absence of colony stimulating factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 6, e26317 (2011).
Greter, M. et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 36, 1031–1046 (2012).
Elmore, M.R. et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397 (2014).
Salter, M.W. & Beggs, S. Sublime microglia: expanding roles for the guardians of the CNS. Cell 158, 15–24 (2014).
Tay, T.L., Savage, J., Hui, C.W., Bisht, K. & Tremblay, M.E. Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J. Physiol. (Lond.) 595, 1929–1945 (2016).
Hong, S., Dissing-Olesen, L. & Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 36, 128–134 (2016).
Davalos, D. et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 (2005).
Nimmerjahn, A., Kirchhoff, F. & Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005).
Abiega, O. et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 14, e1002466 (2016).
Li, Y., Du, X.F., Liu, C.S., Wen, Z.L. & Du, J.L. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev. Cell 23, 1189–1202 (2012).
Fontainhas, A.M. et al. Microglial morphology and dynamic behavior is regulated by ionotropic glutamatergic and GABAergic neurotransmission. PLoS One 6, e15973 (2011).
Eyo, U.B. et al. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J. Neurosci. 34, 10528–10540 (2014).
Dissing-Olesen, L. et al. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci. 34, 10511–10527 (2014).
Tremblay, M.E., Lowery, R.L. & Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8, e1000527 (2010).
Schafer, D.P. et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012).
Paolicelli, R.C. et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).
Lui, H. et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 165, 921–935 (2016).
van Lookeren Campagne, M., Wiesmann, C. & Brown, E.J. Macrophage complement receptors and pathogen clearance. Cell. Microbiol. 9, 2095–2102 (2007).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Marín-Teva, J.L., Cuadros, M.A., Martín-Oliva, D. & Navascués, J. Microglia and neuronal cell death. Neuron Glia Biol. 7, 25–40 (2011).
Brown, G.C. & Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15, 209–216 (2014).
Fourgeaud, L. et al. TAM receptors regulate multiple features of microglial physiology. Nature 532, 240–244 (2016).
Marín-Teva, J.L. et al. Microglia promote the death of developing Purkinje cells. Neuron 41, 535–547 (2004).
Frade, J.M. & Barde, Y.A. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron 20, 35–41 (1998).
Sedel, F., Béchade, C., Vyas, S. & Triller, A. Macrophage-derived tumor necrosis factor alpha, an early developmental signal for motoneuron death. J. Neurosci. 24, 2236–2246 (2004).
Wakselman, S. et al. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 28, 8138–8143 (2008).
Schafer, D.P. & Stevens, B. Microglia function in central nervous system development and plasticity. Cold Spring Harb. Perspect. Biol. 7, a020545 (2015).
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A. & Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 109, E197–E205 (2012).
Bliss, T.V., Collingridge, G.L. & Morris, R.G. Synaptic plasticity in health and disease: introduction and overview. Phil. Trans. R. Soc. Lond. B 369, 20130129 (2013).
Zhao, C., Deng, W. & Gage, F.H. Mechanisms and functional implications of adult neurogenesis. Cell 132, 645–660 (2008).
Akers, K.G. et al. Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science 344, 598–602 (2014).
Rogers, J.T. et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241–16250 (2011).
Schafer, D.P., Lehrman, E.K. & Stevens, B. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia 61, 24–36 (2013).
Koeglsperger, T. et al. Impaired glutamate recycling and GluN2B-mediated neuronal calcium overload in mice lacking TGF-b1 in the CNS. Glia 61, 985–1002 (2013).
George, J., Cunha, R.A., Mulle, C. & Amédée, T. Microglia-derived purines modulate mossy fibre synaptic transmission and plasticity through P2X4 and A1 receptors. Eur. J. Neurosci. 43, 1366–1378 (2016).
Sipe, G.O. et al. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 7, 10905 (2016).
Sierra, A. et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495 (2010).
Bachstetter, A.D. et al. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 32, 2030–2044 (2011).
Gemma, C. & Bachstetter, A.D. The role of microglia in adult hippocampal neurogenesis. Front. Cell. Neurosci. 7, 229 (2013).
Tremblay, M.E. et al. The role of microglia in the healthy brain. J. Neurosci. 31, 16064–16069 (2011).
Parkhurst, C.N. et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013).
Butovsky, O. et al. Identification of a unique TGF-b-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143 (2014).
Sierra, A. et al. The “Big-Bang” for modern glial biology: Translation and comments on Pío del Río-Hortega 1919 series of papers on microglia. Glia 64, 1801–1840 (2016).
Murray, P.J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Ransohoff, R.M. A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991 (2016).
Chiu, I.M. et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Reports 4, 385–401 (2013).
Holtman, I.R. et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol. Commun. 3, 31 (2015).
Pardo, C.A., Vargas, D.L. & Zimmerman, A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 17, 485–495 (2005).
Morgan, J.T. et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 68, 368–376 (2010).
Vargas, D.L., Nascimbene, C., Krishnan, C., Zimmerman, A.W. & Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81 (2005).
Tetreault, N.A. et al. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 42, 2569–2584 (2012).
Gupta, S. et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 5, 5748 (2014).
Suzuki, K. et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 70, 49–58 (2013).
Voineagu, I. et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011).
Schafer, D.P. et al. Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. eLife 5, e15224 (2016).
Derecki, N.C. et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109 (2012).
Maezawa, I. & Jin, L.W. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356 (2010).
Estes, M.L. & McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 353, 772–777 (2016).
Giovanoli, S. et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 339, 1095–1099 (2013).
Patterson, P.H. Maternal infection and immune involvement in autism. Trends Mol. Med. 17, 389–394 (2011).
Garay, P.A., Hsiao, E.Y., Patterson, P.H. & McAllister, A.K. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav. Immun. 31, 54–68 (2013).
Squarzoni, P. et al. Microglia modulate wiring of the embryonic forebrain. Cell Reports 8, 1271–1279 (2014).
Penzes, P., Cahill, M.E., Jones, K.A., VanLeeuwen, J.E. & Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293 (2011).
Feinberg, I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 17, 319–334 (1982–83).
Zhan, Y. et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 17, 400–406 (2014).
Chen, S.K. et al. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell 141, 775–785 (2010).
Paloneva, J. et al. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology 56, 1552–1558 (2001).
Bianchin, M.M. et al. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy–PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell. Mol. Neurobiol. 24, 1–24 (2004).
McCarthy, M.M. Multifaceted origins of sex differences in the brain. Phil. Trans. R. Soc. Lond. B 371, 20150106 (2016).
Bolton, J.L. et al. Gestational exposure to air pollution alters cortical volume, microglial morphology, and microglia–Neuron interactions in a sex-specific manner. Front. Synaptic Neurosci. 9, 10 (2017).
Hanamsagar, R. et al. Generation of a microglial developmental index in mice and in humans reveals a sex difference in maturation and immune reactivity. Glia 65, 1504–1520 (2017).
Hanamsagar, R. & Bilbo, S.D. Sex differences in neurodevelopmental and neurodegenerative disorders: Focus on microglial function and neuroinflammation during development. J. Steroid Biochem. Mol. Biol. 160, 127–133 (2016).
Wyss-Coray, T. & Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2, a006346 (2012).
Lambert, J.C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 45, 1452–1458 (2013).
Zhang, B. et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 153, 707–720 (2013).
Guerreiro, R. et al. TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 (2013).
Jonsson, T. et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 (2013).
Efthymiou, A.G. & Goate, A.M. Late onset Alzheimer's disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12, 43 (2017).
Villegas-Llerena, C., Phillips, A., Garcia-Reitboeck, P., Hardy, J. & Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr. Opin. Neurobiol. 36, 74–81 (2016).
Colonna, M. & Wang, Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201–207 (2016).
Suárez-Calvet, M. et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol. Med. 8, 466–476 (2016).
Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453 (2003).
Kleinberger, G. et al. The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 36, 1837–1853 (2017).
Ulrich, J.D. et al. Altered microglial response to Ab plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 9, 20 (2014).
Jay, T.R. et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 (2015).
Wang, Y. et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071 (2015).
Jay, T.R. et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer's disease. J. Neurosci. 37, 637–647 (2017).
Atagi, Y. et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J. Biol. Chem. 290, 26043–26050 (2015).
Yeh, F.L., Wang, Y., Tom, I., Gonzalez, L.C. & Sheng, M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91, 328–340 (2016).
Kleinberger, G. et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6, 243ra86 (2014).
Suárez-Calvet, M. et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 8, 369ra178 (2016).
Paloneva, J. et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25, 357–361 (2000).
Paloneva, J. et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662 (2002).
Griciuc, A. et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643 (2013).
Bertram, L. et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632 (2008).
Jun, G. et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 67, 1473–1484 (2010).
Fonseca, M.I. et al. Analysis of the putative role of CR1 in Alzheimer's disease: genetic association, expression and function. PLoS One 11, e0149792 (2016).
Terry, R.D. et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580 (1991).
DeKosky, S.T., Scheff, S.W. & Styren, S.D. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 5, 417–421 (1996).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Stephan, A.H., Barres, B.A. & Stevens, B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389 (2012).
Howell, G.R. et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Invest. 121, 1429–1444 (2011).
Williams, P.A. et al. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol. Neurodegener. 11, 26 (2016).
Vasek, M.J. et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543 (2016).
Zamanian, J.L. et al. Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410 (2012).
Sofroniew, M.V. & Vinters, H.V. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35 (2010).
Clarke, L.E. & Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 14, 311–321 (2013).
Chung, W.S., Allen, N.J. & Eroglu, C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb. Perspect. Biol. 7, a020370 (2015).
Liddelow, S.A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Chung, W.S., Welsh, C.A., Barres, B.A. & Stevens, B. Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 18, 1539–1545 (2015).
Iaccarino, H.F. et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235 (2016).
Beggs, S., Trang, T. & Salter, M.W. P2X4R+ microglia drive neuropathic pain. Nat. Neurosci. 15, 1068–1073 (2012).
Sorge, R.E. et al. Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nat. Med. 18, 595–599 (2012).
Masuda, T. et al. Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat. Commun. 7, 12529 (2016).
North, R.A. P2X receptors. Phil. Trans. R. Soc. Lond. B 371, 20150427 (2016).
Tozaki-Saitoh, H. et al. P2Y12 receptors in spinal microglia are required for neuropathic pain after peripheral nerve injury. J. Neurosci. 28, 4949–4956 (2008).
Burnstock, G. Purinergic mechanisms and pain. Adv. Pharmacol. 75, 91–137 (2016).
Guan, Z. et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci. 19, 94–101 (2016).
Masuda, T. et al. Transcription factor IRF5 drives P2X4R+-reactive microglia gating neuropathic pain. Nat. Commun. 5, 3771 (2014).
Masuda, T. et al. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Reports 1, 334–340 (2012).
Tsuda, M. et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424, 778–783 (2003).
Sorge, R.E. et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 18, 1081–1083 (2015).
Ferrini, F. et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl− homeostasis. Nat. Neurosci. 16, 183–192 (2013).
Macosko, E.Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169, 1276–1290.e17 (2017).
Pandya, H. et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 20, 753–759 (2017).
Muffat, J. et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 22, 1358–1367 (2016).
Hammond, T.R. & Stevens, B. Increasing the neurological-disease toolbox using iPSC-derived microglia. Nat. Med. 22, 1206–1207 (2016).
Bohlen, C.J. et al. Diverse requirements for microglial survival, specification, and function revealed by defined-medium cultures. Neuron 94, 759–773.e8 (2017).
Gosselin, D. et al. An environment-dependent transcriptional network specifies human microglia identity. Science 356, eaal3222 (2017).
Choi, S.H. et al. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 515, 274–278 (2014).
Abud, E.M. et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278–293.e9 (2017).
Bennett, M.L. et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 113, E1738–E1746 (2016).
Coull, J.A. et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021 (2005).
Hildebrand, M.E. et al. Potentiation of synaptic GluN2B NMDAR currents by Fyn kinase is gated through BDNF-mediated disinhibition in spinal pain processing. Cell Reports 17, 2753–2765 (2016).
Hodgkin, J. Sex determination and the generation of sexually dimorphic nervous systems. Neuron 6, 177–185 (1991).
Chowen, J.A., Azcoitia, I., Cardona-Gomez, G.P. & Garcia-Segura, L.M. Sex steroids and the brain: lessons from animal studies. J. Pediatr. Endocrinol. Metab. 13, 1045–1066 (2000).
Gill, S.K., Bhattacharya, M., Ferguson, S.S. & Rylett, R.J. Identification of a novel nuclear localization signal common to 69- and 82-kDa human choline acetyltransferase. J. Biol. Chem. 278, 20217–20224 (2003).
Schwarz, J.M., Sholar, P.W. & Bilbo, S.D. Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 120, 948–963 (2012).
Acaz-Fonseca, E., Duran, J.C., Carrero, P., Garcia-Segura, L.M. & Arevalo, M.A. Sex differences in glia reactivity after cortical brain injury. Glia 63, 1966–1981 (2015).
McCullough, L.D. et al. Stroke sensitivity in the aged: sex chromosome complement vs. gonadal hormones. Aging (Albany. NY) 8, 1432–1441 (2016).
Bollinger, J.L., Bergeon Burns, C.M. & Wellman, C.L. Differential effects of stress on microglial cell activation in male and female medial prefrontal cortex. Brain Behav. Immun. 52, 88–97 (2016).
Arevalo, M.A., Santos-Galindo, M., Acaz-Fonseca, E., Azcoitia, I. & Garcia-Segura, L.M. Gonadal hormones and the control of reactive gliosis. Horm. Behav. 63, 216–221 (2013).
Lenz, K.M., Nugent, B.M., Haliyur, R. & McCarthy, M.M. Microglia are essential to masculinization of brain and behavior. J. Neurosci. 33, 2761–2772 (2013).
Stellwagen, D. & Malenka, R.C. Synaptic scaling mediated by glial TNF-a. Nature 440, 1054–1059 (2006).
Shi, Q. et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 9, eaaf6295 (2017).
Fonseca, M.I. et al. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer's disease. J. Neuroinflammation 8, 4 (2011).
Fonseca, M.I., Zhou, J., Botto, M. & Tenner, A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J. Neurosci. 24, 6457–6465 (2004).
Acknowledgements
We thank A. Sengar (Hospital for Sick Children) for important discussions about the review and assistance with the manuscript and figures. Work of the authors is supported by CIHR, Brain Canada, Krembil Foundation (M.W.S.) and NIH, NIH RO1NS071008 (B.S.), Simons Foundation SFARi (B.S.). M.W.S. holds the Northbridge Chair in Paediatric Research at the Hospital for Sick Children.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Salter, M., Stevens, B. Microglia emerge as central players in brain disease. Nat Med 23, 1018–1027 (2017). https://doi.org/10.1038/nm.4397
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm.4397
This article is cited by
-
Differential contribution of THIK-1 K+ channels and P2X7 receptors to ATP-mediated neuroinflammation by human microglia
Journal of Neuroinflammation (2024)
-
The roles of tissue resident macrophages in health and cancer
Experimental Hematology & Oncology (2024)
-
Oxidative stress and inflammation cause auditory system damage via glial cell activation and dysregulated expression of gap junction proteins in an experimental model of styrene-induced oto/neurotoxicity
Journal of Neuroinflammation (2024)
-
Hypoxia inducible factor-1α regulates microglial innate immune memory and the pathology of Parkinson’s disease
Journal of Neuroinflammation (2024)
-
Microglial ferroptotic stress causes non-cell autonomous neuronal death
Molecular Neurodegeneration (2024)
