
Abstract
IMA901 is the first therapeutic vaccine for renal cell cancer (RCC) consisting of multiple tumor-associated peptides (TUMAPs) confirmed to be naturally presented in human cancer tissue. We treated a total of 96 human leukocyte antigen A (HLA-A)*02+ subjects with advanced RCC with IMA901 in two consecutive studies. In the phase 1 study, the T cell responses of the patients to multiple TUMAPs were associated with better disease control and lower numbers of prevaccine forkhead box P3 (FOXP3)+ regulatory T (Treg) cells. The randomized phase 2 trial showed that a single dose of cyclophosphamide reduced the number of Treg cells and confirmed that immune responses to multiple TUMAPs were associated with longer overall survival. Furthermore, among six predefined populations of myeloid-derived suppressor cells, two were prognostic for overall survival, and among over 300 serum biomarkers, we identified apolipoprotein A-I (APOA1) and chemokine (C-C motif) ligand 17 (CCL17) as being predictive for both immune response to IMA901 and overall survival. A randomized phase 3 study to determine the clinical benefit of treatment with IMA901 is ongoing.
Similar content being viewed by others


Main
Therapeutic cancer vaccines hold the promise of combining meaningful efficacy (prolongation of survival) with very good safety and tolerability, as has been shown in several recent randomized trials1,2,3. However, development of cancer vaccines remains a major challenge, with little knowledge of (i) the optimal tumor antigens to target, (ii) suitable agents to counteract regulatory mechanisms opposing successful immunotherapy and (iii) surrogate and predictive biomarkers that can improve our understanding of these regulatory mechanisms and predict a patient's response to therapy.
The first major issue addressed in this work is whether relevant HLA-restricted peptides for immunotherapeutic intervention in patients with RCC can be identified and clinically validated. We defined the relevance of the antigens as their natural presence on the tumor in the majority of RCC samples, their immunogenicity (induction of T cell responses in clinical studies) and the association of the vaccine-induced T cell responses with clinical benefit. For the identification, selection and preclinical immunological validation of such antigens, we used the antigen discovery platform XPRESIDENT4,5 to create a multipeptide vaccine designated IMA901 for immunotherapy of RCC. We tested IMA901 in HLA-A*02+ subjects with advanced RCC in two clinical trials, a phase 1 (n = 28) and a randomized phase 2 (n = 68) trial, both of which assessed the association of T cell responses to IMA901 with clinical benefit.
The second aim was to identify an agent that reduced numbers of Treg cells and that improved the clinical efficacy of the vaccine. A single dose of cyclophosphamide has been used in several previous studies with the intention of inhibiting Treg cells6,7,8,9. However, a beneficial effect of single-dose cyclophosphamide had not previously been conclusively shown in a randomized clinical trial.
The third issue addressed was whether blood cell populations or serum factors could be identified as prognostic or predictive biomarkers. We investigated cell populations known to counteract T cell–based immunotherapy, such as Treg cells10, several populations of myeloid-derived suppressor cells (MDSCs)11,12, interleukin-10 (IL-10)-secreting and IL-17–secreting T cells13 and dysfunctional T cell receptor ζ (TCR-ζ)low or nitrotyrosinehigh T cells14,15, which might have an effect on the prognosis of patients with RCC. Lastly, we also screened >300 serum biomarkers for association with clinical outcome and immune response.
Results
Identification of TUMAPs based on natural presentation
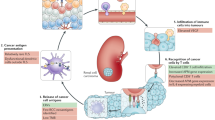
XPRESIDENT is an antigen discovery platform that allows for the identification of thousands of TUMAPs—collectively defined as the HLA ligandome or immunopeptidome—directly from primary tumor tissues (Fig. 1a). It uses a combination of mass spectrometry, gene expression profiling, literature-based functional assessment, in vitro human T cell assays and bioinformatics to select a set of candidate vaccine TUMAPs for the tumor type of interest4,16,17.

(a) Process scheme showing the steps from primary tumor tissues and blood samples to a multipeptide immunotherapeutic cancer vaccine. (b) Shown as an example is the sequencing of the peptide YVDPVITSI (MET-001) encoded by MET and naturally presented by HLA-A*02 on primary RCC tissue by mass spectrometry. m/z, mass to charge ratio; b2–b8, b-series fragment ions; y2–y8, y-series fragment ions. (c) mRNA expression profile of MET. Expression values for bulk normal tissues and several RCC samples are shown relative to normal bulk kidney (with a value arbitrarily set to 1.0). Each bar represents a single microarray measurement. (d) HLA multimer analysis of in vitro–primed T cells generated by stimulation of human CD8+ T cells derived from healthy donors with MET-001 (top) or an irrelevant peptide (bottom) using artificial antigen-presenting cells. The numbers in the quadrants indicate the percentage of CD8+ cells.
We systematically investigated 80 predominantly primary RCC tissues, including 32 evaluable HLA-A*02+ RCC samples, by mass spectrometry, resulting in the identification of peptides from housekeeping and tumor-associated antigens4,18. To identify potentially clinically relevant antigens, we compared the mRNA expression profiles of 42 RCC samples to those of 35 different healthy tissues19 and tested the immunogenicity of candidate tumor-associated antigens determined by gene expression profiling through subsequent in vitro priming using artificial antigen-presenting cells16 to detect antigen-specific T cells in peripheral blood cells of healthy donors. We selected nine HLA-A*02–restricted TUMAPs and one HLA-DR–restricted TUMAP that were derived from highly overexpressed tumor antigens and designated them IMA901 (Table 1). We added an HLA-A*02–restricted peptide from hepatitis B virus (HBV) as a marker peptide for the phase 1 study20 that was not part of the IMA901 formulation in further trials. Data from a representative TUMAP, MET-001, are shown in Figure 1b–d.
Safety, immunogenicity and efficacy in the phase 1 study
To investigate safety and obtain preliminary results on the immunogenicity and potential clinical benefit of IMA901 in humans, we conducted an open-label, multicenter, single-arm phase 1 study in patients with advanced RCC. Subjects received up to eight IMA901 vaccinations, each preceded by administration of granulocyte-macrophage colony-stimulating factor (GM-CSF) as an immunomodulator (Fig. 2a). We enrolled 28 HLA-A*02+ subjects with RCC, 15 of whom had not received prior systemic treatment. The remaining 13 subjects had previously received up to three treatment lines, mostly consisting of cytokines, often in combination with chemotherapy. We observed no treatment-related serious adverse events or deaths during the study period. At a 3-month follow up, disease had progressed in 16 subjects, 11 subjects had stable disease and 1 subject showed a partial response to therapy according to the Response Evaluation Criteria in Solid Tumors (RECIST).

(a) Schedule of vaccinations with IMA901 plus the immunomodulator GM-CSF and of peripheral blood mononuclear cell (PBMC) sampling. Scr, screening; V, vaccination; FU, follow up (no vaccine applied). (b) The observed magnitude of T cell kinetics in a multimer analysis of a representative subject vaccinated with IMA901. The readout antigens were nine HLA class I TUMAPs in one sample (TUMAP pool), the marker peptide HBV-001, which was included in IMA901, and the negative control antigen HIV-001, which was not included in the vaccine. The timeline is shown in weeks, with each tick indicating 1 week. (c) DCR of subjects according to the presence or absence of detectable immune responses (n = 27 subjects total). The numbers above the bars indicate number of subjects with the observed immune responses. Anti-HBV, vaccine-induced response to the HBV peptide. (d) Association of the number of T cell responses with the percentage of Treg cells (n = 26 subjects). Shown on the y axis is the percentage of CD4+FOXP3+ cells among CD45+ lymphocytes in prevaccination samples. Shown on the x axis is the number of vaccine-induced TUMAP responses per subject. Each dot represents an individual subject, and the horizontal lines represent the median values.
Among 27 immune-evaluable subjects, 20 showed a vaccine-induced T cell response to at least one TUMAP and 8 responded to multiple TUMAPs. Fourteen subjects showed a response to the HBV-001 marker peptide. As detailed in the Online Methods, we analyzed responses after a single round of in vitro stimulation and culture to enable the detection of both low and high frequencies of vaccine-induced T cells in the peripheral blood of the subjects. The kinetics of the T cell responses determined at the time points of individual blood drawings before and throughout the course of the vaccinations were analyzed for 13 response courses in 9 subjects in detail and are shown for a representative subject in Figure 2b. We typically observed peak responses at 1–3 weeks after start of vaccinations, and we were still able to detect vaccine-induced immune responses 3 months after start of vaccinations in 7 of 13 response courses.
In a retrospective analysis, we found that subjects who responded to multiple TUMAPs were significantly (P = 0.019) more likely to experience disease control (stable disease or partial response) than subjects who responded to only one TUMAP or had no response (Fig. 2c). In contrast, we found no association between T cell responses to HBV-001 and clinical benefit. To assess whether Treg cells have a role in compromising immune responses in patients, we quantified the number of Treg cells in the subjects before and after vaccination. Low percentages of Treg cells before vaccination were associated with multiple T cell responses to the vaccine in comparison with nonresponders or single TUMAP immune responders (Fig. 2d; P = 0.016).
Safety and efficacy in the phase 2 study
We based the design of the phase 2 study on the observed positive association between disease control and T cell responses to multiple TUMAPs, as well as the negative association of Treg cell numbers with the induction of such responses. We investigated whether an additional immunomodulator (single-dose cyclophosphamide) could improve the efficacy of IMA901 vaccination and the clinical outcome in patients with RCC, presumably by reducing the numbers of Treg cells (Fig. 3a).

Evaluable subjects of the per protocol population are shown in the survival curves. (a) Design of the clinical trial. OS, overall survival; R, randomization; i.d., intradermal administration. (b) PFS of subjects treated with (n = 31) or without (n = 33) cyclophosphamide. (c) Overall survival of subjects treated with (n = 31) or without (n = 33) cyclophosphamide. (d) Overall survival of subjects evaluable for immune responses grouped as follows: immune responders, +Cy arm (n = 17); subjects without an observed immune response, +Cy arm (n = 13); immune responders, −Cy arm (n = 22); subjects without an observed immune response, −Cy arm (n = 9). (e) Overall survival of subjects with no detectable immune responses (n = 22), immune responses to one TUMAP (n = 23), immune responses to two TUMAPs (n = 14) or immune responses to at least three TUMAPs (n = 2).
We randomized 1:1 a total of 68 HLA-A*02+ subjects with metastatic RCC (intention-to-treat population, ITT) with documented progression during or after previous systemic therapy to receive either single-dose cyclophosphamide before the first of 17 vaccinations or no pretreatment (33 subjects comprised the arm that included pretreatment with cyclophosphamide, termed here the '+Cy arm', and 35 subjects comprised the arm with no pretreatment, termed here the '−Cy arm'); 64 subjects were evaluable for the predefined primary efficacy analysis (per protocol). The baseline characteristics and risk factors of the subjects were well balanced overall between the two randomization arms (Supplementary Table 1), except for a shorter median time since diagnosis in the subjects from the +Cy arm (which was reported to be a negative prognostic factor in previous studies21,22).
Confirming the phase 1 results, treatment with IMA901 was safe and generally well tolerated, with mild to moderate local site reactions being the most frequent side effects. Two possibly related serious adverse events were reported, with one subject experiencing a systemic allergic reaction after the twelfth vaccination (caused by GM-CSF, as shown by an in vitro basophil degranulation assay) and another with a grade 3 localized allergic reaction after the eleventh vaccination, with no similar signs of intolerance after further vaccinations. We found no differences in the safety profile between subjects in the +Cy arm and those in the −Cy arm.
Similar to reports for other cancer vaccines, shrinkage of established tumor lesions was infrequent, with one complete response and two partial responses being reported by investigators and one partial response as assessed by centralized review. The disease control rate (DCR; percentage of subjects with complete or partial response or stable disease according to RECIST within all treated subjects), according to a centralized review at 6 months after the start of treatment, was 31% (95% confidence interval (CI) 17–48%) in subjects with prior cytokine treatment and was 14% (95% CI 3–35%) in subjects with prior tyrosine kinase inhibitor (TKI) treatment. Whereas progression-free survival (PFS) was comparable in the two study arms (Fig. 3b), we found a trend for prolonged survival in subjects in the +Cy arm (median overall survival of 23.5 months for +Cy compared with 14.8 months for −Cy, hazard ratio (HR) = 0.57, P = 0.090; Fig. 3c and Supplementary Table 1).
Association of immune responses and overall survival
Of the 64 per protocol subjects, 61 were evaluable for a prospectively defined immune response analysis. The immune response rate of 64% in this group (including 26% multi-TUMAP responders) was similar to that of the phase 1 study (where we investigated more time points). These rates were comparable between subjects with and without cyclophosphamide pretreatment (data not shown), indicating that cyclophosphamide did not alter the induction of T cell responses.
The baseline characteristics of the subjects were generally well balanced between the immune responders and the nonresponders and between multi-TUMAP responders and single-TUMAP responders or nonresponders (Supplementary Table 2). However, age was associated with fewer multi-TUMAP responses (P = 0.01), and there was a tendency toward more responses in male than in female subjects. Neither age nor gender have otherwise been reported as prognostic factors for overall survival of patients with RCC22.
Despite the observed similarity of prognostic factors in the subjects in the +Cy and −Cy arms, a prospectively defined analysis showed that among immune responders, subjects had prolonged survival if pretreated with cyclophosphamide compared with subjects without this pretreatment (HR = 0.38, P = 0.040) (Fig. 3d). In contrast, we found no difference in survival of subjects in the +Cy and −Cy arms among nonimmune responders (HR = 0.92, P = 0.870). Although these data are based on small numbers of subjects because of the trial design, they suggest that a single dose of cyclophosphamide does not have antitumor activity by itself but instead supports the effects of the vaccine as an immunomodulator and enables the translation of immune responses into clinical benefit. Furthermore, survival time was extended if a subject had a response to multiple TUMAPs (P = 0.023 log-rank test for trend in subjects with increasing number of responses) (Fig. 3e), corroborating the observation from the phase 1 study that clinical benefit is associated with the breadth of vaccine-induced immune responses.
Downmodulation of Treg cells by cyclophosphamide
To assess the effects of single-dose cyclophosphamide on the numbers of Treg cells in patients with RCC, we characterized these cells23,24 as CD45+CD3+CD4+CD8−FOXP3+CD25hiCD127low by immunophenotyping. A prospectively defined analysis showed an approximately 20% reduction in Treg cell numbers 3 d after compared with immediately before the cyclophosphamide infusion in the +Cy arm (P = 0.013); we found no such effect in the −Cy arm and no effect on absolute lymphocyte counts in subjects from either arm (Fig. 4a).

(a) The medians of the absolute Treg cell and lymphocyte counts of evaluable phase 2 subjects at different visits: VB, screening up to 4 weeks before V1; VC, administration of single-dose cyclophosphamide 3 d before V1; V1, first IMA901 vaccination; V6, sixth vaccination 3 weeks after V1. Treg cells of evaluable ITT subjects pretreated with cyclophosphamide (n = 25 at VC, n = 29 at V1, n = 28 at V6), Treg cells of subjects not pretreated with cyclophosphamide (n = 31 at VC, n = 27 at V1, n = 27 at V6), lymphocytes of subjects pretreated with cyclophosphamide (n = 33 at VB, n = 31 at VC, n = 33 at V1, n = 32 at V6) and lymphocytes of subjects not pretreated with cyclophosphamide (n = 35 at VB, n = 35 at V1, n = 34 at V6) are shown. The decrease in the numbers of Treg cells from VC to V1 was significant in the +Cy arm (P = 0.013 by paired two-sided Wilcoxon test, n = 24). (b,c) Phenotype of circulating Treg cells in evaluable subjects treated with cyclophosphamide (b) (n = 23 at VC, n = 29 at V1) or not treated with cyclophosphamide (c) (n = 31 at VC, n = 27 at V1). Frequency of Ki-67+ cells among the Treg cells was significantly decreased from VC to V1 in subjects treated with cyclophosphamide (P = 0.0006 by paired two-sided Wilcoxon test, n = 23). Each dot represents an individual subject, and the horizontal lines represent the median values.
Furthermore, quantifying the expression of Ki-67 showed that the percentage of proliferating cells among all Treg cells was decreased 3 d after compared with before cyclophosphamide treatment (Fig. 4b,c).
Assessment of cellular biomarkers before treatment
To establish a profile of the immune-regulatory environment of patients with RCC, we analyzed different cellular biomarkers in samples from subjects of the phase 2 study obtained before treatment with cyclophosphamide and IMA901 and from matched healthy controls. MDSCs are myeloid cells with immunosuppressive properties that have been proposed to negatively modulate anticancer immunity. We developed a panel of antibodies to identify six MDSC phenotypes in a single multicolor staining: MDSC1 (CD14+, interleukin-4 receptor α (IL-4Rα)+)25, MDSC2 (CD15+IL-4Rα+)25, MDSC3 (Lineage−HLA-DR−CD33+)26,27, MDSC4 (CD14+HLA-DR−/lo)28, MDSC5 (CD11b+CD14−CD15+)14 and MDSC6 (CD15+FSCloSSChi)29. We found that the percentage of MDSC2–MDSC6 phenotypes among all lymphocytes were significantly (P < 0.01) higher in subjects with RCC than in the controls (Fig. 5a and Supplementary Fig. 1a–e).

(a–e) Comparison of cellular biomarkers of evaluable ITT subjects aged <70 years (n = 50–53 per group) and age- and gender-matched healthy controls (n = 22). Each dot represents an individual subject, and horizontal lines represent the median values. Statistical assessments were performed using unpaired two-sided Wilcoxon tests. MDSC4, P = 0.0043 (a); TCR-ζ, P < 0.0001 (b); nitrotyrosine expression of T cells, P = 0.0038 (c); IL-17+, P < 0.0001 (d); IL-10+, P < 0.0001 (e). (f) Overall survival of evaluable per protocol subjects having high (n = 29) or low (n = 28) MDSC4 numbers; HR = 0.35, P = 0.0035 by log-rank test. (g–j) Overall survival in evaluable per protocol subjects having high or low concentrations of APOA1 (g) or CCL17 (i); subgroup analyses were performed with respect to cyclophosphamide pretreatment (h,j). The HR for overall survival in APOA1high compared with APOA1low subjects was 0.41 (P = 0.007 by log-rank test, n = 31 compared with n = 30, respectively), 0.26 in the +Cy arm (P = 0.006, n = 16 compared with n = 14) and 0.62 in the −Cy arm (P = 0.278, n = 15 compared with n = 16). The HR for overall survival in CCL17high compared with CCL17low subjects was 0.41 (P = 0.011 by log-rank test, n = 30 compared with n = 30, respectively), 0.19 in the +Cy arm (P = 0.003, n = 14 compared with n = 16) and 0.69 in the −Cy arm (P = 0.418, n = 16 compared with n = 14).
Two key mechanisms by which MDSCs cause T cell dysfunction have previously been reported: depletion of arginine, which induces T cell receptor ζ chain downregulation14, and the generation of reactive oxygen species, which induces T cell tyrosine nitration30,31. Indeed, we found that TCR-ζ expression and nitrotyrosine expression by T cells—both measured as median fluorescence—were significantly lower (P < 0.0001) and higher (P = 0.0038), respectively, in patients compared with controls (Fig. 5b,c). TCR-ζ expression was significantly inversely correlated with numbers of MDSC2–MDSC4 (P < 0.05, data not shown) and strongly inversely correlated with MDSC5 (P < 0.0001; data not shown). We found no association between nitrotyrosine expression and the numbers of phenotypes MDSC1–MDSC6 (data not shown).
IL-17+CD4+ T cells (T helper type 17 (TH17) cells) represent a distinct lineage of helper T cells with proinflammatory effector functions. TH17 cells have been found to be elevated in several human cancers13,32,33 and may predict metastatic progression13. Comparison of patients with RCC and controls (Fig. 5d,e and Supplementary Fig. 1f,g) showed that TH17 cells were significantly enriched in the peripheral blood of subjects with RCC (P < 0.0001; Fig. 5d). Furthermore, the concentrations of IL-10+CD4+ T cells were also significantly higher in patients with RCC than in controls (P < 0.0001, Fig. 5e).
In a retrospective analysis, the numbers of two out of the six MDSC phenotypes were significantly negatively associated with overall survival: MDSC4 (P < 0.001 by continuous analysis; patients with high compared with low numbers of MDSC4 are shown in Fig. 5f) and MDSC5 (P = 0.016). TCR-ζ expression tended to associate positively with survival (P = 0.084).
Assessment of serum biomarkers before treatment
To identify serum parameters that could predict a potentially increased immunogenicity and overall survival by IMA901 treatment, we measured >300 analytes in samples from subjects in the IMA901 phase 2 study before treatment. To distinguish prognostic biomarkers (that is, those associated with clinical outcome independent of therapy) from predictive biomarkers, we considered only those biomarkers that were associated with both immune response and with overall survival in the +Cy arm but not in the −Cy arm. The use of this method was justified by the observation that immune responses to TUMAPs were associated with clinical benefit in both clinical studies described here (whereas responses to HBV-001 were not) and by the finding that subjects from the +Cy arm showed longer overall survival times (HR = 0.57, P = 0.090) and better association of immune response with overall survival compared with subjects in the −Cy arm.
Post-hoc univariate statistical analyses revealed that serum concentrations of two analytes, APOA1 and CCL17, were positively predictive for immune responses (P = 0.016 and P = 0.032, respectively) and multipeptide responses (P < 0.0001 and P = 0.0028, respectively; data not shown). High concentrations of APOA1 and CCL17 were able to identify patient populations with significantly longer overall survival (P < 0.007 and P < 0.011, respectively), with this effect being pronounced in the +Cy arm but absent in the −Cy arm (Fig. 5g–j).
Discussion
To the best of our knowledge, this work is the first to show that HLA-restricted peptides, selected by virtue of being naturally presented by cancer, elicit specific vaccine-induced immune responses that are associated with clinical benefit. Most therapeutic vaccine trials in cancer so far have not shown such an association, which may be explained by the lack of confirmation of natural presentation on patient-derived primary tumor tissue. In two trials testing IMA901, the breadth of the T cell response (that is, the number of different specific antigens against which T cell responses were induced by the vaccine) was significantly associated with clinical benefit, implying that the targeting of multiple antigens in immunotherapy is required for clinical efficacy. The finding that immune responses are associated with clinical benefit could be explained by therapeutic efficacy or by immune responses being merely a prognostic factor. Three observations argue in favor of the hypothesis that vaccine-induced immune responses are indeed causally related to better clinical outcome: (i) the association of immune response with clinical benefit was only observed for tumor-associated peptides and not for the unrelated nontumor HBV-derived peptide; (ii) immune responses were associated with survival only in subjects randomized to a single dose of cyclophosphamide; and (iii) the baseline characteristics were overall well matched between multipeptide immune responders, immune responders and nonresponders (Supplementary Table 2), and the two observed differences (age and sex) are not considered prognostic factors in advanced RCC.
Moreover, this is the first randomized trial to show that single-dose cyclophosphamide is associated with prolonged survival after tumor immunotherapy. This effect is only observed in subjects with specific immune responses to IMA901, indicating that single-dose cyclophosphamide does not have any single-agent activity (either as a cytotoxic or as a nonspecific immunomodulatory drug) but rather acts as an immunomodulator of the vaccine. Differences in baseline factors cannot explain this better outcome; if anything, subjects pretreated with cyclophosphamide tended to have a slightly worse prognosis, as exemplified by the shorter time from tumor diagnosis to randomization in this group21,22. The results also show that cyclophosphamide predominantly affects proliferating Treg cells within the Treg population. A prior report suggested that peripheral Treg cells have a higher fraction of proliferating cells than do other cell subsets34.
The survival of patients who received cytokine treatment prior to IMA901 vaccination plus cyclophosphamide (with the median overall survival not reached after 33.1 months) compared favorably with data recently published from studies with TKIs in the same setting (median overall survival of 17.8 months for sorafenib treatment compared with 15.2 months for placebo35 and 16.4 months for sunitinib treatment36), whereas the overall survival of patients receiving IMA901 vaccination without cyclophosphamide (median overall survival of 15.8 months) matched these trials quite well. The baseline characteristics and risk factors of the subjects in the IMA901 phase 2 trial were generally comparable in this inter-trial comparison (Supplementary Table 3). Although encouraging, this observation must be interpreted with caution, as it was derived from a comparison with historical controls.
Our work also suggests that, in addition to Treg cells, MDSCs may have a role in RCC outcome. Among several MDSC phenotypes described in the literature, only two populations, here termed MDSC4 and MDSC5, were shown to be negatively associated with survival in patients with RCC. Similar cellular populations have been shown to be prognostic in breast and colon cancer37. This observation is new for RCC and for cancer under immunotherapy. The TKI sunitinib, a standard first-line treatment in advanced RCC, has been shown to decrease the numbers of Treg cells in mice38 and patients with RCC39, as well as MDSCs in patients with RCC40. Based on this rationale, a randomized phase 3 trial combining IMA901 with single-dose cyclophosphamide and sunitinib compared with treatment with sunitinib alone was started recently41.
The retrospective screening of a large number of serum biomarkers and their association with several outcome variables showed that two biomarkers, APOA1 and CCL17, are potentially predictive for vaccine-induced immunity and overall survival. APOA1 is the major protein component of high-density lipoprotein. Reduced concentrations of high-density lipoprotein and APOA1 are found in several types of cancer42 and are associated with chronic inflammatory and oxidative stress, which may suppress adaptive immune responses43. CCL17 is a chemoattractant for several immune cells, including CD4+ and CD8+ T cells, in particular, skin-homing44,45 and tumor-infiltrating T cells46. A major source of CCL17 is myeloid dendritic cells; constitutively as well as induced production of CCL17 has a role in dendritic cell function47,48. Based on these exploratory biomarker results, we hypothesize that high serum concentrations of APOA1 and CCL17 may predict immune and clinical response to IMA901.
Several aspects of the study reported here strongly suggest that therapeutic vaccinations with IMA901 induce specific immune responses that lead to clinical benefit and may be predicted by a defined pretreatment biomarker pattern. However, relatively small numbers of patients have been evaluated thus far, requiring further confirmation of clinical benefit as a prospectively defined endpoint in a large randomized clinical trial, which is currently ongoing. This randomized trial will also serve to validate the proposed predictive biomarker signature in a prospectively defined fashion.
Finally, we conclude that the rational clinical development of therapeutic cancer vaccines accompanied and guided by a comprehensive biomarker program—including T cell response monitoring, as well as monitoring of other cellular and serum biomarkers—is feasible even in multicenter trials and is valuable, provided that sample quality is tightly controlled. Such biomarkers reflect relevant mechanisms of immune induction and tolerance and assist in defining patient subpopulations with a higher prospect of response to cancer immunotherapy. These findings may have relevance for other cancer immunotherapy trials.
Methods
Phase 1 study design.
Between 2005 and 2006, we conducted a multicenter, single-arm, open-label first-in-man study at six study sites in three European countries in accordance with the Declaration of Helsinki, current International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidelines and all applicable regulatory and ethical requirements. All subjects provided written informed consent before study-related procedures were performed. Subjects with histologically confirmed RCC of American Joint Committee on Cancer Stages 3 and 4 were enrolled.
Each of the vaccinations consisted of an intradermal (i.d.) injection of GM-CSF (75 μg) followed within 15–30 min by an i.d. injection of IMA901 (413 μg of each peptide). The vaccine therapy was a monotherapy, meaning that no other antitumor therapies were concomitantly administered during the study course. Of the 28 HLA-A*02–positive patients, 27 patients received eight vaccinations and 1 patient was prematurely withdrawn and received only one vaccination.
The primary endpoint was safety and tolerability. Main secondary endpoints were immunogenicity and evidence of antitumor activity.
Phase 2 study design and treatment schedule.
Between 2007 and 2009, we conducted a multicenter, randomized, open-label phase 2 study in accordance with the Declaration of Helsinki, current ICH guidelines and all applicable regulatory and ethical requirements. All subjects provided written informed consent before study-related procedures were performed. Subjects were recruited by 23 centers across ten European countries. Subjects were randomized to receive or not receive one single infusion of cyclophosphamide (300 mg m−2) 3 d before the first vaccination with IMA901 plus GM-CSF. The phase 2 injection doses of IMA901 per peptide and GM-CSF were identical to the phase 1 study, however formulation in the phase 2 study excluded the marker peptide HBV-001. Stratification factors included risk group (Memorial Sloan-Kettering Cancer Center (MSKCC) three-item score, with only favorable and intermediate risk patients being eligible) and previous treatment (cytokine or TKI). Subjects had to have measurable lesions to be eligible for the trial; other relevant inclusion criteria were documented tumor progression during or after previous systemic therapy and Karnofsky performance status ≥80%. Subjects with brain metastases, relevant autoimmune diseases, other malignancies or major abnormalities of hematology, liver and renal function tests were excluded. No treatment with either anticancer agents or immunosuppressants was allowed within 4 weeks before entering the trial. Subjects in both treatment arms were to receive seven vaccinations in the first 5 weeks of treatment (induction period) followed by ten further vaccinations at 3 week intervals for up to 30 weeks (maintenance period). Subjects were prospectively defined as per protocol if they received at least six vaccinations with IMA901 and GM-CSF.
The primary endpoint was DCR after 6 months. Main secondary endpoints were PFS, overall survival, immunogenicity and safety.
Clinical assessment criteria.
The safety of study treatment was assessed in both trials based on the occurrence of adverse events, which were categorized and graded according to National Cancer Institute Common Toxicity Criteria (NCI-CTC).
Evidence of antitumor activity was obtained in terms of tumor size of target lesions and evaluation of nontarget lesions at baseline (that is, during the screening period) and at the respective follow-up visits as per RECIST 1.0.
In the phase 1 study, tumor follow-up assessment was compared with baseline. In the phase 2 study, tumor assessments were performed every 6 weeks (except for the first assessment, which occurred 8.5 weeks after cyclophosphamide administration). Tumor images were centrally reviewed by independent radiologists and one oncologist (Perceptive Informatics Inc., Berlin, Germany). Further follow up to collect survival data was conducted at 3-month intervals. The phase 2 trial is registered and has ClinicalTrials.gov identifier NCT01265901. The ongoing phase 3 trial, which is mentioned in the Discussion section, has the ClinicalTrials.gov identifier NCT00523159.
Immunomonitoring and biomarker analysis.
PBMCs were isolated from subjects within 8 h of venipuncture using fully standardized procedures, cryopreserved and shipped for central laboratory analysis. We detected T cell responses to individual HLA-A*02–binding peptides by performing HLA multimer staining and interferon γ (IFN-γ) enzyme-linked immunospot (ELISPOT) after antigen stimulation and 12 d of in vitro culture. We assessed the numbers of Treg cells and MDSCs and the expressions of TCR-ζ and nitrotyrosine on T cells by ex vivo multicolor flow cytometric analyses. We performed assessment of TH subset numbers by intracellular cytokine staining after a 5 h stimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin. We measured serum biomarkers using multiplex technologies, mass spectrometry and ELISA.
Statistical analyses.
In the phase 1 study, we analyzed the association of immune responses with DCR using a two-sided χ2 test and the association of Treg cells with immune responses retrospectively using a two-sided Wilcoxon test.
In the phase 2 study, we performed prospectively defined binary survival comparisons of study subgroups using log-rank tests and deduced survival rates from Kaplan-Meyer plots. We indicated 95% confidence intervals in the text. In the case of multiple immune response groups, the log-rank test for trend was used testing the null hypothesis that there is no linear trend between increasing numbers of immune responses and median survival.
We assessed the decrease of absolute Treg cell numbers from VC to V1 with a prospectively defined paired two-sided Wilcoxon test and used the same test in the post-hoc analysis of Ki-67+ cells within Treg cells.
In further post-hoc analyses, we used an unpaired two-sided Wilcoxon test to compare the number of MDSCs, TH subset frequencies and expressions of age- and gender-matched patients and healthy controls. We performed correlation analyses among cellular biomarkers post hoc using a Pearson product-moment correlation.
We performed all serum biomarker analyses retrospectively: an unpaired Welch's t test was used for association of serum biomarkers with T cell responses. We assessed the association of continuous biomarker parameters with overall survival in a univariate Cox proportional hazards model. To use continuous parameters as classifiers distinguishing between subjects with more and less survival benefit, we used the median concentrations derived from the per protocol population as a cutoff. The log-rank test was performed for binary survival comparisons.
We performed statistical analyses using R (GNU) programming language using the package 'survival', GraphPad Prism 4 (GraphPad Software) and Statgraphics Centurion (Statpoint Technologies).
Identification and gene expression analysis of HLA-restricted peptides.
After approval by the relevant Institutional Review Boards at the University of Tübingen and Kyoto Prefectural University of Medicine and informed consents given by the patients, HLA peptide pools from shock-frozen tissue samples were obtained by immune precipitation from solid tissues using the HLA-A*02–specific antibody BB7.2 (ref. 59) for HLA-A*02+ patients (BB7.2 was kindly provided by H.-G.R., antibody amounts used for precipitation were adopted to the weight of each tissue and ranged from 10–25 mg). Peptides were sequenced using a quadrupole time of flight (Q-TOF) mass spectrometer, as described elsewhere18, or separated using a nanoAcquity UPLC system (Waters, Eschborn, Germany) and analyzed in an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). All selected sequences were verified by comparison with the spectra of synthetic peptides (data not shown). Gene expression analysis was performed using high-density oligonucleotide microarrays, as previously described18.
RCC samples included samples from European and Japanese patients and had to fulfill standardized quality criteria to be evaluable. No relevant differences in mRNA expression or peptide presentation of the TUMAPs contained in IMA901 between the European and Japanese patients were observed. IMA901 TUMAPs were found on HLA-A*0201+ as well as on HLA-A*0206+ tumor tissues (data not shown).
In vitro validation of TUMAP immunogenicity.
In vitro T cell priming using artificial antigen-presenting cells loaded with high-density HLA-peptide complexes and antibodies to CD28 was performed as described previously16.
Peptide vaccine preparation.
All individual peptides and the final multipeptide vaccine IMA901 have been manufactured in compliance with good manufacturing practice and in accordance to predefined release specifications. Peptides were manufactured with purities ≥95.0% (Bachem AG, Bubendorf, Switzerland) with a free N terminus, free C terminus and acetate or hydrochloride as a counter ion using standard solid-phase procedures. IMA901 was formulated (Rentschler Biotechnologie GmbH, Laupheim, Germany), labeled, packaged and distributed to clinical sites (Penn Pharmaceutical Services, Tredegar, UK). IMA901 for clinical phase 1 was formulated by mixing solutions of the 11 peptides, as listed in Table 1, lyophilised without the addition of excipients and resolubized with 4.2% sodium bicarbonate buffer (Braun AG, Melsungen, Germany). For clinical phase 2, the formulation of IMA901 comprised all peptides also used for phase 1, with the exception of HBV-001, with added mannitol (Merck KGaA, Darmstadt, Germany) and poloxamer 188 (Lutrol F68) (BASF Chemtrade GmbH, Burgbernheim, Germany) as excipients. Addition of these excipients did not alter the immunological properties of the vaccine, as shown in preclinical vaccination studies. Briefly, C57BL/6 mice were immunized with the ovalbumin-derived peptide SIINFEKL and CpG deoxyoligonucleotide as an immunomodulator in 4.2% bicarbonate buffer with or without poloxamer 188 and mannitol added at concentrations identical to those in the later formulation of IMA901 for the phase 2 trial. Mice were killed 9 d after immunization, and the frequency of peptide-specific CD8+ T cells was estimated. There was no difference in immune responses between the groups with and without the excipients (data not shown).
Quality control of the identity of the specified parameters, the content of active ingredients, the uniformity of content, the purity and the product-related impurities of the formulated product included the implementation of a validated analytical HPLC method, which was able to separate each individual peptide. Using this method, stability studies of the lyophilized product did not show any substantial decrease in peptide content or in peptide purity (product-related impurities) when the vaccine was stored at 25 °C (60% relative humidity), 5 °C and −20 °C for up to 36 months (data not shown). In accordance with the stability data obtained after reconstitution, the peptide vaccine IMA901 had to be used within 1 h after reconstitution on clinical sites.
Immunomodulators.
The immunomodulators GM-CSF (sargramostim, Leukine; Genzyme, Seattle, USA) and cyclophosphamide (Endoxan; Baxter, UK) were purchased as good manufacturing practice grade lyophilisates and administered i.d. or intravenously, respectively, according to the manufacturer's instructions.
PBMC sample acquisition.
PBMCs were isolated from 50 ml (phase 1) or 100 ml (phase 2) sodium heparine blood within 8 h of venipuncture using a standard Ficoll-Hypaque density gradient procedure and were cryopreserved in serum-free medium. At sites where this was feasible, cells were stored in the gas phase of liquid nitrogen before shipment; otherwise cells were directly shipped within 7 d of storage at −80 °C. After shipment to the central laboratory on dry ice, frozen cells were stored in the gas phase of liquid nitrogen until use. All media and reagents were standardized, tested and supplied centrally as kits. All crucial steps of PBMC sample acquisition were performed according to standard operating procedures by specifically trained personnel. Laboratories isolating PBMCs had to pass a documented qualification procedure, including the successful performance of test runs.
Discussion of the choice of assay for detection of T cell responses.
The T cell assays performed routinely for patients in the IMA901 phase 1 and phase 2 studies were 'in vitro stimulation assays', which involve a 12-d cell culture after peptide pulsing. This contrasts to the quantification of T cells 'ex vivo' without prior cell culture but does not constitute 'in vitro priming' of naive T cells, for which professional antigen-presenting cells have to be used in vitro (see Fig. 1d for an example of in vitro priming).
We justify this choice based on a number of observations and further assumptions. (i) For a subset of six patients of the IMA901 phase 1 study, for whom ex vivo IFN-γ ELISPOT assays were performed, no patients showed ex vivo–measurable vaccine-induced immune responses, whereas four showed a measurable IFN-γ ELISPOT response after in vitro stimulation. (ii) The amplification factor of the in vitro stimulation assay was ∼100× based on direct comparisons (ex vivo compared with in vitro stimulated) of virus-specific T cell responses (n = 33), as well as strong patient vaccine-induced responses (n = 3; data not shown). Based on this observation, it can be assumed that the majority of the observed T cell responses of IMA901 phase 1 and phase 2 patients were well below the detection threshold of current ex vivo multimer assays (∼0.01%). (iii) The empirical threshold for detection of a vaccine-induced response was set to a conservative value (4× above baseline), as the in vitro culture is expected to be associated with a resulting lower precision as compared with an ex vivo assay. (iv) In several reported immunotherapy studies60,61,62,63,64, ex vivo detectable, vaccine-induced T cell responses in patients with melanoma were readily detected. It is worth noting that in these studies, altered ligands or the HLA-A*02-restricted Melan-A peptide were almost exclusively used. It is expected that for altered, nonself antigens, higher precursor frequencies of naive T cells as compared with self antigens can be found. For the particular Melan-A peptide used, an abnormally high precursor frequency of naive T cells has been shown65. However, the biological relevance of altered ligands as vaccine targets is subject to debate66, as high-frequency T cells specific to the altered peptide may not crossreact to the unaltered peptide that is presented by the tumor.
We conclude that for the detection of vaccine-induced responses to self antigens in patients with cancer, in vitro stimulation assays are the current technical state of the art, as such responses are often below the detection limit of current ex vivo assays. The low-frequency responses in the peripheral blood that are detected by the in vitro (but missed by the ex vivo) assay may be biologically relevant, as indicated by the phase 1 and 2 IMA901 studies and other studies. This biological relevance of low-frequency responses in the blood may be explained by higher T cell frequencies in other compartments (for example, lymph nodes, bone marrow or tumor sites) or association with epitope spreading to other targets.
Assessment of T cell responses.
Vaccine-induced T cell responses to HLA-A*02 binding peptides were identified by HLA multimer staining or IFN-γ ELISPOT after one 12-d in vitro stimulation of samples of patients with peptides from single or pooled blood drawings, as specified below. For all patients, both assays were performed, and patients had to be evaluable for at least one assay for each peptide at each visit during which immunomonitoring was scheduled. Because of the technical limitations of the assays, no readout was available for the single HLA-DR–associated peptide. In both assays, time point pools were used from the limited amount of blood to derive a full dataset for all HLA class I peptides contained in IMA901 plus the controls (phase 1: VC/V1 (pooled), V4/V5, V6/V7, V8/FU; phase 2: VC/V1, V5, V6, V7). Additionally, PBMC samples for each single time point were analyzed by multimer staining in a subset of nine patients with 13 detected responses in the phase 1 study to investigate kinetics of T cell responses.
For each analysis time point, approximately 1.2 × 107 thawed PBMCs in a concentration of 2.5 × 106 to 5.4 × 106 ml−1 were incubated in X-Vivo 15 plus 2 mM L-glutamine (Lonza, Verviers, Belgium or Life Technologies, Darmstadt, Germany) containing all IMA901 class I peptides used for immunization (10 μg ml−1 per peptide) for 2 h at 37 °C, 5% CO2 and 95% humidity. Cells were pelleted, resuspended in X-Vivo 15 supplemented with 10% heat-inactivated human AB Serum (C.C. Pro, Neustadt/W., Germany or PAN, Aidenbach, Germany), 2 mM l-glutamine, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin (Lonza) and 20 U ml−1 IL-2 (Novartis, Munich, Germany) and cultured for 12 d. Half of the medium was replaced every 2–3 d.
ELISPOT assay.
The IFN-γ ELISPOT assessment was performed using the ELISPOT-IFN-γ Set and the AEC Substrate Reagent Set (both by Becton Dickinson, Heidelberg, Germany), according to the manufacturer's protocol. For stimulations, PBMCs were harvested on day 13, washed in CTL-Wash (CTL, Cleveland, USA), supplemented with 2 mM L-glutamine and re-stimulated for readout in CTL-Test (CTL) supplemented with 2 mM L-glutamine. For each peptide assessment, 75,000 effector cells per well were incubated with 100,000 untreated K562-A2 cells67 (phase 1) or 75,000 K562-A2 cells67 pretreated with 100 U ml−1 human IFN-γ (Boehringer Ingelheim, Ingelheim, Germany) (phase 2) and 10 μg ml−1 of peptide for 20–24 h at 37 °C and 5% CO2. Readout was performed on a Series 3B ELISPOT Reader (CTL). Each sample was tested in triplicate, and the presence of vaccine-induced response was determined according to objective criteria based on suggestions by the international Association for Cancer Immunotherapy (CIMT) Immunoguiding Program (CIP) and others68,69. Briefly, the mean spot number for a given peptide had to be at least twofold over the mean background spot number, and the difference of the triplicates had to pass the Student's t test (P < 0.05, two sided). Furthermore, the ratio of peptide to negative control mean for a given post-vaccination time point had to be at least twofold over the corresponding ratio before vaccination.
HLA multimer assay.
Dual HLA multimer assessment was performed as shown previously70 with minor modifications. Briefly, PBMCs were harvested on day 13 and approximately 0.5 × 106 to 1 × 106 PBMCs per staining were treated first with HLA-A*0201 multimer phycoerythrin and HLA-A*0201 multimer allophycocyanin (individually at 5 μg ml−1 HLA concentration plus all multimers in one pool each at 1 μg ml−1) (phase 1) or LIVE/DEAD fixable Aqua dead cell stain kit (Life Technologies) followed by multimers (phase 2) and then by antibodies to CD8 (SK1, 1:20, BD) plus antibodies to CD3 (UCHT1, 1:40, BD). Cells were fixed and analyzed on a FACSCalibur (phase 1, BD) or LSRII SORP cytometer (phase 2, BD). FCSExpress (DeNovo Software, Los Angeles, USA) or FlowJo (Tree Star, Ashland, USA) was used to enumerate single multimer-positive cells among CD8+CD3+ lymphocytes. Detailed gating and representative dot plots are shown in Supplementary Figures 2 and 3. The presence of a vaccine-induced response was determined according to objective criteria based on suggestions by the international Association for Cancer Immunotherapy (CIMT) Immunoguiding Program (CIP) and others68,69. Briefly, frequencies of multimer-positive cells must have been at least fourfold over the corresponding frequency before vaccination. Furthermore, in dot plots, multimer-positive cell populations must have been clustered and discrete from multimer-negative cells. In phase 2, this was determined by the evaluation of blinded data by a jury consisting of five members. The jury members had to evaluate whether the multimer-stained cell population was clearly clustered and discrete. Each jury member could attribute up to two points, and a vaccine-induced immune response was only identified if at least nine points were given. Cells must have been specifically stained with one single HLA multimer to exclude particles nonspecifically binding to all HLA multimers.
Assessment of Treg cells.
PBMC samples were thawed in the presence of 0.5 U ml−1 Benzonase (Merck, Darmstadt, Germany).
In phase 1, quantification of Treg cells was performed according to the manufacturers' protocol of the FOXP3 (PCH101, 1:5) staining set and antibody to CD4 (L3T4, 1:4) (both obtained from eBioscience, San Diego, USA) and CD45 (2D1, 1:4, BD). The sample analysis was performed on a FACSCalibur cytometer, and data analysis was performed using FCSExpress. All Treg cell measurements were performed in one single assay, and quantification was based on an automatically adjusted percentile quadrant (99.7% FOXP3 percentile on CD4−CD45+ lymphocytes). The results using CD4, FOXP3 and CD45 were later confirmed with a selected subset of patient samples by a four-color cytometric analysis that included CD25 (BC96, 1:5; data not shown).
In phase 2, quantification of Treg cells among PBMCs was performed by the LIVE/DEAD fixable Aqua dead cell stain kit followed by surface staining for CD45 (F10-89-4, 1:00, AbD Serotec, Düsseldorf, Germany), CD4 (S3.5, 1:600) and CD8 (3B5, 1:400, both Life Technologies), CD3 (SK7, 1:80), CD25 (M-A251, 1:5), CD127 (human IL-7R–M21, 1:20) and CD45RA (L48, 1:240, all from BD). Erythrocytes were lysed with Pharm-Lyse-Solution (BD), and cells were fixed and permeabilized (FOXP3 staining buffer set, eBiosciences) followed by intracellular staining for FOXP3 (206D, 1:8, Biolegend, Fell, Germany) and Ki-67 (B56, 1:10, BD). Fluorochromes were carefully chosen to optimize the signal-to-noise ratio on the instrument platform according to criteria described before71. After fixation with 1% formaldehyde, cells were analyzed on a BD LSRII SORP cytometer, and data analysis was performed using FlowJo (TreeStar). Identical gates (Supplementary Fig. 4) were established for all samples and in all assays with the help of fluorescence minus one (FMO) and isotype controls that clearly identified the outlines of the populations. Samples were set evaluable if at least 75,000 live CD45+ lymphocytes were counted. To assess Treg cell kinetics data for individual patients, a total of seven assays were performed, and the data were pooled. To control the interassay variability of the Treg cell assays, ten additional unique healthy donor control samples were thawed and stained in each experiment. Data bridging between assays was ensured by assay standardization, calibrated instrument settings, standardized data evaluation procedures and comparison with internal PBMC controls.
To assess the precision of the assay, several analyses were performed. First, the evaluation of the ten unique control samples that were repeated in each of the phase 2 Treg cells assays described above resulted in a mean interassay coefficient of variation (CV) of 13%. In an independent validation study, testing a panel of 13 different healthy donor samples resulted in a mean interassay CV of 7.1%. The large majority (58 of 60) of patient kinetics were evaluated from assays containing all time points of a patient, and, therefore, the (generally lower) intra-assay CV should be applied for these analyses. From the analysis of replicate samples within one assay, we determined in one study an intra-assay CV of 9.6% and in another study an intra-assay CV of 4.4%.
Absolute Treg cell frequencies were calculated by multiplying relative Treg cell numbers within CD45+ lymphocytes measured by flow cytometry with absolute lymphocyte counts from hematology. Hematology was not performed at VC for the −Cy arm. Therefore, absolute Treg cell numbers for VC in the −Cy arm were calculated with lymphocyte counts from V1 (3 d after VC).
Matched comparisons of cellular biomarkers between patients and healthy donors.
PBMCs of healthy blood bank donors (Institute of Clinical and Experimental Transfusion Medicine, Tübingen) were obtained after informed consent from all subjects and approval of the protocol by the local Institutional Review Board at the University of Tübingen. For comparison of pretreatment concentrations with healthy donors, ITT patients below the age of 70 were selected. The resulting groups of patients and controls were balanced with respect to age, gender and cytomegalovirus seropositivity.
Assessment of MDSC frequencies, TCR-ζ and nitrotyrosine expression.
PBMC samples were thawed in the presence of 0.5 U ml−1 Benzonase (Merck).
For the assessment of MDSCs, cells were first stained with the LIVE/DEAD fixable Aqua dead cell stain kit. Cells were then blocked with human IgG (AbD Serotec) to reduce subsequent Fc-mediated binding. Cells were then surface stained with antibodies to CD45 (HI30, 1:100, Life Technologies), antibodies to CD15 (MMA, 1:80), CD11b (ICRF44, 1:640), CD124 (human IL-4R–M57, 1:10), CD14 (MϕP9, 1:60), CD33 (P.67.6, 1:320), Lineage marker (consisting of CD3 (UCHT1, 1:40), CD14 (M5E2, 1:40) and CD19 (HIB19, 1:320) and CD56 (NCAM16.2, 1:80, all by BD)). Erythrocyte lysis and fixation was performed with FACS lysing solution (BD).
For the assessment of TCR-ζ and nitrotyrosine, cells were first stained with the LIVE/DEAD fixable Aqua dead cell stain kit. Cells were then surface stained with antibodies to CD4 (S3.5, 1:300), CD8 (3B5, 1:400, both Life Technologies), CD45 (F10-89-4, 1:100, AbD) and CD3 (SK7, 1:160, BD). Stained cells were lysed for erythrocytes (Pharm-Lyse-Solution, BD), permeabilized and fixed (eBioscience). After intracellular staining with antibodies to CD3-ζ (1A6, 1:20, Santa Cruz Biotechnology, Santa Cruz, USA) and antibodies to nitrotyrosine (6B10.2, 1:50, Merck Millipore, Schwalbach, Germany), cells were again fixed with formaldehyde (Fluka, Munich, Germany).
Fluorochromes of the multicolor assays were carefully chosen to optimize the signal-to-noise ratio on the instrument platform according to criteria described before71. Measurement was performed on an LSR II SORP instrument (BD). All samples were analyzed in one experiment for each assay type. The data analysis was performed with FlowJo (TreeStar). Identical gates were used for all samples (Supplementary Fig. 5). Samples were set evaluable if at least 75,000 live CD45+ lymphocytes were counted.
Assessment of TH cell subsets.
After thawing, PBMCs were stimulated for 5 h with 50 ng ml−1 PMA (Sigma Aldrich, Munich, Germany) and 750 ng ml−1 ionomycin (Merck) at 37 °C. After treatment with GAMUNEX (Bayer, Leverkusen, Germany) and ethidium monoazide (Life Technologies) for 10 min on ice, cells were stained for CD3 (UCHT1, 1:250, Life Technologies), CD4 (RPA-T4, 1:250, BioLegend), CD8 (SK1, 1:25), CCR4 (1G1, 1:15) and CCR6 (11A9, 1:25, all BD). Cells were then permeabilized with a Cytofix/Cytoperm kit (BD) and stained with antibodies to IL-17 (eBio64DEC17, 1:5, eBiosciences), tumor necrosis factor α (TNF-α) (cA2, 1:10), IL-10 (B-T10, 1:6, Miltenyi Biotech, Bergisch Gladbach, Germany) and IFN-γ (B27, 1:250, BioLegend). Cells were analyzed immediately on an LSR II cytometer with FACSDiva software (BD Biosciences). In all experiments at least 1 × 106 cells were measured. One control donor was always included in each experiment. Flow cytometry staining and data analysis were performed on blinded patient samples.
Serum biomarker sample collection.
All serum samples were collected at visit VC. Samples were taken using a serum/gel-VACUTAINER 5 ml (BD), inverted and incubated for a minimum of 30 min. Tubes were centrifuged for 15 min at ≥1,200 g, and serum (approximately 2 ml) was transferred into a NUNC cryotube (3.6 ml). Cryotubes were stored immediately at or below −20 °C until measurement.
Serum biomarker analyses.
More than 300 serum analytes (cytokines, chemokines, other proteins and metabolites) were measured using various multiplex technologies, mass spectrometry and ELISA.
APOA1 was measured by Rules Based Medicine (RBM, Austin, TX) as part of a multiplex assay using luminex technology (Human MAP v1.6). CCL17 measurement was part of a tenplex cytokine panel provided by Millipore, and the assay was performed in the Naturwissenschaftliches und Medizinisches Institut (NMI) in Reutlingen, Germany.
References
Kantoff, P.W. et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 28, 1099–1105 (2010).
Schwartzentruber, D.J. et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 364, 2119–2127 (2011).
Schuster, S.J. et al. Vaccination with patient-specific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J. Clin. Oncol. 29, 2787–2794 (2011).
Weinschenk, T. et al. Integrated functional genomics approach for the design of patient-individual antitumor vaccines. Cancer Res. 62, 5818–5827 (2002).
Singh-Jasuja, H., Emmerich, N.P. & Rammensee, H.G. The Tubingen approach: identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol. Immunother. 53, 187–195 (2004).
Ghiringhelli, F. et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 56, 641–648 (2007).
Motoyoshi, Y. et al. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol. Rep. 16, 141–146 (2006).
Ikezawa, Y. et al. Cyclophosphamide decreases the number, percentage and the function of CD25+ CD4+ regulatory T cells, which suppress induction of contact hypersensitivity. J. Dermatol. Sci. 39, 105–112 (2005).
Lutsiak, M.E. et al. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 105, 2862–2868 (2005).
Campbell, D.J. & Koch, M.A. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nat. Rev. Immunol. 11, 119–130 (2011).
Peranzoni, E. et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 22, 238–244 (2010).
Gabrilovich, D.I. & Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174 (2009).
Derhovanessian, E. et al. Pretreatment frequency of circulating IL-17+CD4+ T-cells, but not Tregs, correlates with clinical response to whole-cell vaccination in prostate cancer patients. Int. J. Cancer 125, 1372–1379 (2009).
Zea, A.H. et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 65, 3044–3048 (2005).
Nagaraj, S., Schrum, A.G., Cho, H.I., Celis, E. & Gabrilovich, D.I. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J. Immunol. 184, 3106–3116 (2010).
Walter, S. et al. Cutting edge: predetermined avidity of human CD8 T cells expanded on calibrated MHC/anti-CD28–coated microspheres. J. Immunol. 171, 4974–4978 (2003).
Rammensee, H.G., Weinschenk, T., Gouttefangeas, C. & Stevanovic, S. Towards patient-specific tumor antigen selection for vaccination. Immunol. Rev. 188, 164–176 (2002).
Krüger, T. et al. Lessons to be learned from primary renal cell carcinomas: novel tumor antigens and HLA ligands for immunotherapy. Cancer Immunol. Immunother. 54, 826–836 (2005).
Mathiassen, S., Lauemoller, S.L., Ruhwald, M., Claesson, M.H. & Buus, S. Tumor-associated antigens identified by mRNA expression profiling induce protective anti-tumor immunity. Eur. J. Immunol. 31, 1239–1246 (2001).
Bertoletti, A. et al. Molecular features of the hepatitis B virus nucleocapsid T-cell epitope 18–27: interaction with HLA and T-cell receptor. Hepatology 26, 1027–1034 (1997).
Motzer, R.J., Bacik, J., Murphy, B.A., Russo, P. & Mazumdar, M. Interferon-α as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. J. Clin. Oncol. 20, 289–296 (2002).
Heng, D.Y. et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J. Clin. Oncol. 27, 5794–5799 (2009).
Liu, W. et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 203, 1701–1711 (2006).
Seddiki, N. et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J. Exp. Med. 203, 1693–1700 (2006).
Mandruzzato, S. et al. IL4Rα+ myeloid-derived suppressor cell expansion in cancer patients. J. Immunol. 182, 6562–6568 (2009).
Kusmartsev, S. et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 14, 8270–8278 (2008).
Mirza, N. et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 66, 9299–9307 (2006).
Filipazzi, P. et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J. Clin. Oncol. 25, 2546–2553 (2007).
Schmielau, J. & Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res. 61, 4756–4760 (2001).
Bronte, V. et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J. Exp. Med. 201, 1257–1268 (2005).
Nagaraj, S. et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 13, 828–835 (2007).
Zhang, B. et al. The prevalence of Th17 cells in patients with gastric cancer. Biochem. Biophys. Res. Commun. 374, 533–537 (2008).
Kryczek, I. et al. Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment. J. Immunol. 178, 6730–6733 (2007).
Tuovinen, H. et al. Thymic production of human FOXP3+ regulatory T cells is stable but does not correlate with peripheral FOXP3 expression. Immunol. Lett. 117, 146–153 (2008).
Escudier, B. et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 356, 125–134 (2007).
Motzer, R.J. et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 24, 16–24 (2006).
Solito, S. et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 118, 2254–2265 (2011).
Hipp, M.M. et al. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood 111, 5610–5620 (2008).
Finke, J.H. et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin. Cancer Res. 14, 6674–6682 (2008).
Ko, J.S. et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 15, 2148–2157 (2009).
Rini, B.I. et al. IMA901 Multipeptide Vaccine Randomized International Phase III Trial (IMPRINT): a randomized, controlled study investigating IMA901 multipeptide cancer vaccine in patients receiving sunitinib as first-line therapy for advanced/metastatic RCC. J. Clin. Oncol. 29 (suppl.15), abstr. TPS183 (2011).
Muntoni, S. et al. Serum lipoproteins and cancer. Nutr. Metab. Cardiovasc. Dis. 19, 218–225 (2009).
Burger, D. & Dayer, J.M. High-density lipoprotein-associated apolipoprotein A-I: the missing link between infection and chronic inflammation? Autoimmun. Rev. 1, 111–117 (2002).
Andrew, D.P. et al. C–C chemokine receptor 4 expression defines a major subset of circulating nonintestinal memory T cells of both Th1 and Th2 potential. J. Immunol. 166, 103–111 (2001).
Bromley, S.K., Mempel, T.R. & Luster, A.D. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat. Immunol. 9, 970–980 (2008).
Kanagawa, N. et al. CC-chemokine ligand 17 gene therapy induces tumor regression through augmentation of tumor-infiltrating immune cells in a murine model of preexisting CT26 colon carcinoma. Int. J. Cancer 121, 2013–2022 (2007).
Real, E. et al. Immature dendritic cells (DCs) use chemokines and intercellular adhesion molecule (ICAM)-1, but not DC-specific ICAM-3–grabbing nonintegrin, to stimulate CD4+ T cells in the absence of exogenous antigen. J. Immunol. 173, 50–60 (2004).
Semmling, V. et al. Alternative cross-priming through CCL17–CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat. Immunol. 11, 313–320 (2010).
Schmidt, S.M. et al. Induction of adipophilin-specific cytotoxic T lymphocytes using a novel HLA-A2–binding peptide that mediates tumor cell lysis. Cancer Res. 64, 1164–1170 (2004).
Sadovnikova, E., Jopling, L.A., Soo, K.S. & Stauss, H.J. Generation of human tumor-reactive cytotoxic T cells against peptides presented by non-self HLA class I molecules. Eur. J. Immunol. 28, 193–200 (1998).
Schag, K. et al. Identification of C-met oncogene as a broadly expressed tumor-associated antigen recognized by cytotoxic T-lymphocytes. Clin. Cancer Res. 10, 3658–3666 (2004).
Brossart, P. et al. Identification of HLA-A2–restricted T-cell epitopes derived from the MUC1 tumor antigen for broadly applicable vaccine therapies. Blood 93, 4309–4317 (1999).
Brossart, P. et al. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood 96, 3102–3108 (2000).
Brossart, P. et al. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 61, 6846–6850 (2001).
Wierecky, J. et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res. 66, 5910–5918 (2006).
Boss, C.N. et al. Identification and characterization of T-cell epitopes deduced from RGS5, a novel broadly expressed tumor antigen. Clin. Cancer Res. 13, 3347–3355 (2007).
Dengjel, J. et al. Unexpected abundance of HLA class II presented peptides in primary renal cell carcinomas. Clin. Cancer Res. 12, 4163–4170 (2006).
Ruppert, J. et al. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell 74, 929–937 (1993).
Parham, P. & Brodsky, F.M. Partial purification and some properties of BB7.2. A cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum. Immunol. 3, 277–299 (1981).
Kirkwood, J.M. et al. Immunogenicity and antitumor effects of vaccination with peptide vaccine+/−granulocyte-monocyte colony-stimulating factor and/or IFN-α2b in advanced metastatic melanoma: Eastern Cooperative Oncology Group Phase II Trial E1696. Clin. Cancer Res. 15, 1443–1451 (2009).
Baumgaertner, P. et al. Ex vivo detectable human CD8 T-cell responses to cancer-testis antigens. Cancer Res. 66, 1912–1916 (2006).
Liénard, D. et al. Ex vivo detectable activation of Melan-A–specific T cells correlating with inflammatory skin reactions in melanoma patients vaccinated with peptides in IFA. Cancer Immun. 4, 4 (2004).
Speiser, D.E. et al. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J. Clin. Invest. 115, 739–746 (2005).
Walker, E.B. et al. gp100(209–2M) peptide immunization of human lymphocyte antigen-A2+ stage I–III melanoma patients induces significant increase in antigen-specific effector and long-term memory CD8+ T cells. Clin. Cancer Res. 10, 668–680 (2004).
Pittet, M.J. et al. High frequencies of naive Melan-A/MART-1–specific CD8+ T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med. 190, 705–715 (1999).
Speiser, D.E. et al. Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc. Natl. Acad. Sci. USA 105, 3849–3854 (2008).
Britten, C.M. et al. The use of HLA-A*0201–transfected K562 as standard antigen-presenting cells for CD8+ T lymphocytes in IFN-γ ELISPOT assays. J. Immunol. Methods 259, 95–110 (2002).
Britten, C.M. et al. The CIMT-monitoring panel: a two-step approach to harmonize the enumeration of antigen-specific CD8+ T lymphocytes by structural and functional assays. Cancer Immunol. Immunother. 57, 289–302 (2008).
Chianese-Bullock, K.A. et al. MAGE-A1-, MAGE-A10-, and gp100-derived peptides are immunogenic when combined with granulocyte-macrophage colony-stimulating factor and montanide ISA-51 adjuvant and administered as part of a multipeptide vaccine for melanoma. J. Immunol. 174, 3080–3086 (2005).
Walter, S. et al. High frequencies of functionally impaired cytokeratin 18-specific CD8+ T cells in healthy HLA-A2+ donors. Eur. J. Immunol. 35, 2876–2885 (2005).
Mahnke, Y.D. & Roederer, M. Optimizing a multicolor immunophenotyping assay. Clin. Lab. Med. 27, 469–85 (2007).
Acknowledgements
We thank all investigators and patients participating in the clinical studies. Samples of healthy donors were kindly provided by D. Wernet (Institute of Clinical and Experimental Transfusion Medicine, Tübingen). The cell line K562-A2 was kindly provided by C. Britten (Tumorvakzinezentrum Mainz). This work was supported by the German 'Förderprogramm Biotechnologie Baden-Württemberg' projects 720.830-1-1/46b and 720.830-3-9, the German 'Fortüne' project 1862-0-0, the German 'Deutsche Forschungsgemeinschaft Sonderforschungsbereich 685' projects A6, C5 and C9, the German 'Bundesministerium für Bildung und Forschung' project 0315890F, the Italian 'Fondazione Cassa di Risparmio di Verona, Vicenza, Belluno e Ancona' and the Italian 'Associazione Italiana Ricerca sul Cancro' (grant 6599). We thank H. Schild for providing critical input to the manuscript and the members of the Immatics' Scientific Advisory Board and the Data Safety Monitoring Board for critical input throughout the development program.
Author information
Authors and Affiliations
Contributions
S.W. designed and supervised the immunomonitoring and cellular biomarker program. T.W. designed and supervised the antigen discovery and the noncellular biomarker program. T.W. and H.S.-J. designed the composition of the drug. A.S. was the principal investigator of the IMA901 phase 1 and 2 studies and participated in the discovery phase. A.S., M.S. and P.-Y.D. treated a substantial number of patients in the phase 1 study. R.Z., A.P. and C.S. treated a substantial number of patients in the phase 2 study. W.B. was involved in the discovery phase and immunomonitoring and recruited patients in the phase 2 study. R.M., N.H. and D.M. performed and contributed essentially to the immunomonitoring. R.M. and N.H. performed and contributed essentially to the cellular biomarker program. N.H. and O.S. contributed essentially to the discovery phase and design of the clinical trials. J. Fritsche performed the bioinformatics analyses. A.M. and J. Fritsche performed and contributed essentially to the noncellular biomarker program. V.V. and C.T. supervised and performed experiments in the discovery phase. P.L. and C.F. supervised and performed the pharmaceutical development, respectively. H.P. and J.J.S. contributed essentially to the PBMC preparations for immunomonitoring. V.B., S.M., G.P. and E.D. were essentially involved in the design and data analysis of the cellular biomarker program. E.D. and T.B. performed some of the cellular biomarker experiments. H.Y., T.M., F.H., N.T., K.H. and H.T. were involved in the discovery phase. H.-G.R. and S.S. contributed essentially to the discovery phase and to the design of the overall treatment approach. H.-G.R. contributed essentially to the design of the clinical phase 1 and 2 studies. A.M.-M., H.S.-J. and J. Frisch designed the phase 1 study and planned the analysis. A.M.-M. and J. Frisch supervised the conduct and the analysis of the phase 1 study. A.K., J. Frisch and H.S.-J. designed the phase 2 study and planned the analysis. A.K. and J. Frisch supervised the conduct of the phase 2 study. A.K. and C.R. supervised the analysis of the phase 2 study. S.W., H.S.-J., T.W. and C.R. wrote the manuscript. G.P., V.B. and H.-G.R. provided critical input in the writing of the manuscript. J. Frisch and C.R. had the overall medical oversight. H.S.-J. had the overall scientific oversight. H.-G.R. provided the conceptual basis for the work presented here.
Corresponding author
Ethics declarations
Competing interests
S.W., T.W., R.M., N.H., O.S., J. Fritsche, A.M., D.M., V.V., C.T., P.L., C.F., J. Frisch, A.M.-M., A.K., C.R. and H.S.-J. have a competing interest as employees of Immatics Biotechnologies GmbH and as owners of stock options, shares or both.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5 and Supplementary Tables 1–3 (PDF 3327 kb)
Rights and permissions
About this article
Cite this article
Walter, S., Weinschenk, T., Stenzl, A. et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med 18, 1254–1261 (2012). https://doi.org/10.1038/nm.2883
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm.2883
This article is cited by
-
A Phase I-II multicenter trial with Avelumab plus autologous dendritic cell vaccine in pre-treated mismatch repair-proficient (MSS) metastatic colorectal cancer patients; GEMCAD 1602 study
Cancer Immunology, Immunotherapy (2023)
-
Tumor-infiltrating CD8+ T cells recognize a heterogeneously expressed functional neoantigen in clear cell renal cell carcinoma
Cancer Immunology, Immunotherapy (2022)
-
Landscape of Myeloid-derived Suppressor Cell in Tumor Immunotherapy
Biomarker Research (2021)
-
Targeting macrophages in cancer immunotherapy
Signal Transduction and Targeted Therapy (2021)
-
Beyond conventional immune-checkpoint inhibition — novel immunotherapies for renal cell carcinoma
Nature Reviews Clinical Oncology (2021)
