
Abstract
CD8+ T cells play an important role in immunity to viruses. Just how important these cells are is demonstrated by the evolution of viral strategies for blocking the generation or display of peptide–major histocompatibility complex class I complexes on the surfaces of virus-infected cells. Here, we focus on viral interference with antigen presentation; in particular we consider the importance (and difficulty) of establishing the evolutionary significance (that is, the ability to enhance viral transmission) of viral gene products that interfere with antigen presentation in vitro.
Similar content being viewed by others


Main
Viruses are the ultimate obligate intracellular parasites. Lacking virtually all the machinery necessary for their own replication, they consist of a fragment of nucleic acid (encoding anywhere from less than ten to several hundred proteins) enclosed in a protein or proteolipid shell with just enough of the right polymerases to initiate gene expression (“bad news wrapped in protein”, according to Peter Medawar). For viruses to survive in nature, they must devise a means to be transmitted between hosts. This, not dissemination within a host, is their sole evolutionary selection factor, although dissemination is usually a prerequisite for transmission. At a bare minimum, viruses must encode information to enable regeneration of their structural proteins. Because hosts are generally hostile to sharing their resources with viruses, viruses need to be a bit cleverer than this.
Virus versus host: role of CD8 + T cells
Through various mechanisms, host cells may sense the presence of a virus and attempt to trigger apoptosis to preclude viral replication. Viruses can counter these attempts (see Review by Ware and colleagues in this issue). The presence of unusual nucleic acids produced in the early stages of viral replication (for example, double-stranded RNA) triggers protein kinase R (PKR), which induces an antiviral state in the cells and synthesis and release of interferons (IFNs). IFNs signal the presence of virus to the cellular immune system, whose initial response consists of natural killer (NK) cells. Some viruses have devised means for evading NK cell recognition (see Review by Strominger and colleagues in this issue).
The second wave of the cellular immune counterattack consists of CD8+ T cells, which are activated via the presentation of viral antigens by professional antigen-presenting cells (APCs). The contribution of CD8+ T cells to antiviral immunity has been extensively demonstrated in mouse model systems. Starting with experiments in the late 1960s, T cells (which were later shown to be CD8+) were demonstrated to be responsible for the recovery of mice from acute mousepox (ectromelia) infection1,2,3. Subsequently, we have learned that CD8+ T cells are important in mouse immunity to many viruses. CD8+ T cells exert antiviral effects via the localized secretion of molecules in close vicinity to the virus-infected APC (professional or not). Many CD8+ T cells kill APCs by releasing perforin and granzymes (if APCs express Fas, engagement by Fas ligand on CD8+ T cells can also induce lysis). In addition, nearly all CD8+ T cells secrete IFN-γ and tumor necrosis factor-α (TNF-α) (a small percentage of cells secrete IFN-γ only), which induces a potent antiviral state in cells.
It is important to emphasize that CD8+ T cells are but one of many weapons deployed by the immune system to combat viruses. NK cells, CD4+ T cells and antibody all participate in antiviral immunity. Mice deficient in CD8+ T cell responses effectively handle infections with many viruses, although they do succumb to others, such as ectromelia. Direct demonstration of the role of CD8+ T cells in human antiviral immunity is more difficult to establish. On the one hand, adoptively transferred virus-specific CD8+ T cells are effective against human cytomegalovirus (HCMV)4, HIV-15 and Epstein-Barr virus (EBV)6. On the other hand, T cell–deficient individuals seem to do quite well with common viral infections. There is a single hereditary condition that selectively interferes with CD8+ T cell induction: absence of functional transporter associated with antigen processing (TAP) (described below). Curiously, afflicted individuals suffer from bacterial, not viral, infections7. These patients show only a partial reduction in CD8+ T cells, however, and the residual CD8+ T cell activity may be sufficient to handle common viral infections. Evolutionary selection for individual elements of the immune system may be punctuated: for example, the sporadic appearance of a potentially lethal pathogen would be sufficient to maintain an immune effector mechanism that reduces its lethality. It is plausible, for example, that variola virus (the agent of smallpox) would display a greatly increased mortality rate in TAP-deficient individuals.
Perhaps the best evidence for the importance of CD8+ T cells in human immunity to viruses is the lengths that some human viruses have gone to interfere with antigen presentation, which is the principal focus of our review.
Antigen-presentation primer
To enhance nonspecialists' comprehension of the area of antigen processing and presentation we shall briefly discuss how viral proteins are processed for recognition by virus-specific CD8+ T cells8,9. The specificity of CD8+ T cells is conferred by the clonally restricted T cell receptor (TCR) that interacts with residues from both major histocompatibility complex (MHC) class I molecules and an oligopeptide (>90% are 8–11 amino acids in length) encoded by a viral gene. Peptides are predominantly generated from the byproducts of proteasomal degradation (Fig. 1). Most of the substrates consist of defective ribosomal products (DRiPs) that are degraded within 30 min of their synthesis10. This process enables the rapid recognition of viral proteins early in infections, when viral proteins represent a tiny fraction of total cell proteins. Peptides are transported into the endoplasmic reticulum (ER) by the TAP protein. Here, MHC class I molecules are folded through the concerted actions of general purpose molecular chaperones working with a dedicated chaperone (tapasin) that tethers MHC class I to TAP. Upon peptide binding, MHC class I molecules dissociate from TAP, leave the ER and make their way to the plasma membrane via the Golgi complex.

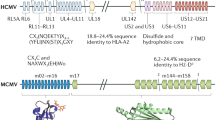
Oligopeptides (red circle) are derived from DRiPs through the action of proteasomes. Nascent MHC class I molecules—consisting of a heavy chain and β2-microglobulin (β2M)—bind to TAP via tapasin. Peptide binding releases MHC class I to the cell surface. VIPRs interfere with this process at multiple steps. (1) EBV EBNA-1 contains a sequence that renders it resistant to proteasomal degradation23,60. HCMV IE is phosphorylated by another viral protein, preventing generation of the immunodominant peptide epitope30. (2A) HSV ICP4761,62 and a BHV1 protein63 bind to the cytosolic side of TAP and prevent peptide translocation. (2B) HCMV US6 binds to TAP in the ER lumen and prevents peptide translocation64. (2C) Several VIPRs bind to MHC class I in the ER, retaining it and/or interfering with the function of the peptide-loading complex. The proteins include AdE319K (retains MHC class I and also prevents tapasin-mediated docking with TAP65); HCMV US3 (binds MHC class I but dissociates, the mechanism of MHC class I retention is not clear but may involve repeated rebinding by newly synthesized US3)66,67. MCMV m4/gp34 forms extensive complexes with MHC class I in the ER68. (3) HCMV US2 and US11 and MHV-68 K3 bind MHC class I in the ER and induce retrotranslocation for degradation by the proteasome69,70. For US11, ubiquitylation of heavy chain cytoplasmic tail, possibly after retrograde translocation, has been described53. MHV-68 K3 ubiquitylates the tail before translocation71. HIV-1 Vpu also induces degradation of newly synthesized MHC class I72—probably via retrograde translocation and proteasome degradation—although, unlike US2 and US11, proteasome activity is required for retrograde translocation73. (4) MCMV m152/gp40 causes MHC class I to be retained in the ER cis-Golgi complex intermediate compartment (ERGIC)74. The fact that m152, itself a distant MHC class I homolog, can reduce cell surface expression of H-60 as well as classical MHC class I (in an allomorph-specific manner75) makes its poorly understood mechanism of action of particular interest. Only the lumenal domain of m152/gp40 is required for the retention of MHC class I76. Binding of m152/gp40 to MHC class I has not been demonstrated, and m152/gp40 itself has a default pathway of export to and degradation in the lysosome74. (5) Several VIPRs remove MHC class I either from the Golgi or the cell surface. MCMV m6/gp48 contains a lysosomal targeting di-leucine motif in its tail; it binds directly to MHC class I and redirects it to the lysosome77. HIV and SIV Nef use the PACS-1 (phosphofurin acidic cluster sorting protein 1)–sorting protein to remove MHC class I from the cell surface and sequester it in the trans-Golgi network78. KSHV K3 and K5 ubiquitylate the MHC class I tail and target MHC class I to lysosomes in a TG101-dependent manner54. K5 also targets ICAM-1 (intercellular adhesion molecule 1) and B7-279. (6) MCMV m4/gp34 is found associated with MHC class I at the cell surface80 and inhibits CTL recognition75, but cause and effect have not been firmly linked.
As peptide-MHC (pMHC) class I complexes accumulate at the cell surface, they have a greater chance of triggering activation by CD8+ T cells with a cognate receptor. The half-life of cell surface complexes depends on the koff of the bound peptide; immunogenic peptides generally dissociate with half-lives in the range of hours to hundreds of hours. There is no simple relationship between complex stability or abundance and the magnitude of the cognate CD8+ T cell response11.
It is important to distinguish naïve CD8+ T cells from armed effector CD8+ T cells. Naïve CD8+ T cells can only be activated by cells expressing the proper costimulatory molecules, that is, by professional APCs. Dendritic cells (DCs) are believed to be the principal APCs for activating naïve CD8+ T cells, although the evidence is largely circumstantial. Effector CD8+ T cells have no such limitation and require only recognition of a cognate complex for activation, although expression of adhesion molecules can decrease the number of complexes required. Effector CD8+ T cells circulate “ready to kill” with preformed perforin and granzymes, but only express IFN-γ and TNF-α upon activation by interaction with a virus-infected APC.
Viral strategies for circumventing CD8 + T cells
In principal, viruses can thwart CD8+ T cell function by interfering with the activation of naïve cells, CD8+ T cells trafficking to infected cells, antigen presentation to effector CD8+ T cells or CD8+ T cell–mediated effects on virus-infected cells. For viruses that are capable of infecting CD8+ T cells, destroying responding CD8+ T cells would seem to be a simple strategy. This mechanism has been described in vitro (but not demonstrated in vivo) for herpes simplex virus (HSV)12. Viruses with this ability are highly unusual, which is interesting, inasmuch as it suggests that hosts are capable of modulating the antiviral state in a cell type–specific manner, with CD8+ T cells being particularly difficult to infect. Blocking activation of naïve CD8+ T cells can be achieved by preventing APC antigen presentation. Many viruses are capable of infecting DCs, and there have been a number of reports that viruses interfere with DC-mediated CD8+ T cells activation in vitro13,14,15,16. This is an area, however, with a potentially enormous gap between in vitro phenomenology and in vivo reality. There is no certain relationship between DCs propagated ex vivo and immature DCs residing in the tissue or activated DCs that have migrated to draining lymph nodes once activated by viral infection. Viral infections generally result in a gross alteration of host cell functions over time. The conditions of infection in vivo and in vitro can differ in terms of the number of viral particles that initiate the infection. Even if a virus-induced decline in antigen presentation measured in vitro accurately reflects the in vivo situation, there could be sufficient residual presentation to enable CD8+ T cell activation in vivo. For example, whereas the antigen-presenting function of DCs in vitro is adversely affected by infection with vaccinia virus (VV)15, VV-infected DCs can be visualized presenting antigens to CD8+ T cells in lymph nodes; this provides the most direct evidence, to date, that DCs function as APCs in viral infections17.
Numerous poxviruses and herpesviruses encode molecules with known or likely effects on chemokine function. As chemokines direct trafficking of CD8+ T cells to the sites of inflammation, these could influence the effectiveness of CD8+ T cells. A secreted chemokine ligand homolog, murine herpes virus 68 (MHV-68) M3, has been implicated as affecting CD8+ T cell function18. Many viruses induce an anti-apoptotic state that should hinder the cytolytic activity of CD8+ T cells. Resistance to the effects of IFN-γ and TNF-α is also a common viral strategy19,20.
Viruses block the presentation of endogenous antigens by APCs by expressing VIPRs (viral proteins interfering with antigen presentation, pronounced “viper”, with an etymological nod to herpesviruses). Numerous VIPRs have been described, and they target virtually all steps in the antigen-processing and -presentation pathway (Fig. 1). This appears to be the principal means by which viruses interfere with CD8+ T cell function, and it is certainly the best characterized. For these reasons we will focus on VIPRs, but not their cell biology and mechanisms of function, which have been the subject of detailed reviews19,20,21. Instead, we will deal primarily with the in vivo function of VIPRs, a topic ripe for review.
Cross-priming: a hurdle to VIPR function
Viruses that block CD8+ T cell activation by expressing VIPRs in APCs face a potentially insurmountable hurdle in cross-priming22. In this process, DCs (and macrophages) internalize antigen from infected cells and reprocess it in the cytosol or endosomal compartment for presentation on their own MHC class I molecules. The nature of the cross-priming material is unknown at present, but it is likely to represent a mixture of phagocytosed intact dying cells and cell debris containing intact and partially degraded proteins, some complexed with sundry molecular chaperones. Because, by definition, cross-priming APCs are not infected, viruses can only impose global restrictions on antigen presentation by releasing soluble inhibitory factors from infected cells. In lieu of this, the best they can do is to evolve proteins that cannot be processed by cross-priming, but even this appears to be rather difficult. EBV nuclear antigen 1 (EBNA-1) is not processed and presented by infected cells due to its indigestibility by proteasomes23, yet EBNA-1–specific CD8+ T cells appear to be generated in vivo by cross-priming24.
Cross-priming is a robust process that may have evolved in response to viral interference with CD8+ T cell activation. There is now solid evidence for cross-priming in a number of viral systems25,26,27,28. Ultimately, the most compelling evidence for the effectiveness of cross-priming may be the vigorous responses to viruses that are loaded with genes encoding proteins capable of interfering with the generation of pMHC class I complexes. A murine CMV (MCMV) determinant whose presentation was completely abrogated by m152 in vitro was equally immunodominant in mice infected with wild-type virus or m152-deficient viruses29. This strongly suggests that cross-priming contributes to the generation of CD8+ T cells specific for this determinant. Similarly, the enigma that CD8+ T cells specific for immediate early protein (IE) are readily generated in HCMV-infected individuals when antigen presentation of IE is efficiently prevented in HCMV-infected cells30 is probably explained by cross-priming. Cross-priming can occur by either the classical cytosolic processing route or via the alternative endosomal pathway whose details are just beginning to appear from the mists31. An important question regarding the endosomal route is the extent to which the peptides it generates overlap with those generated by the cytosolic route, particularly because distinct proteases are involved.
Herpesviruses: a VIPR catalog
Although known VIPRs are encoded by retroviruses and adenoviruses, herpesviruses are the clear champions in the evolution of VIPRs. Herpesviruses encode proteins that interfere with virtually every step of antigen processing and presentation (Fig. 1); indeed, individual family members encode multiple VIPRs (HCMV encodes at least four). It seems worthwhile to pause to consider the characteristics of this family of viruses that are so hell-bent on interfering with CD8+ T cell function.
Herpesviruses are large double-stranded DNA viruses that replicate their genomes with high fidelity compared to RNA viruses (for example, HIV or influenza virus). Herpesviruses fall into three subfamilies. α-herpesviruses are neurotropic: HSV and varicella zoster (VZV, the cause of chickenpox and shingles) are the prototypes. β-herpesviruses are ubiquitous, highly species-specific and cause minimal or no disease in immunocompetent hosts. They include CMVs and human herpesvirus 6 (HHV6) and HHV7. γ-herpesviruses fall into two subgroups: the γ1 prototype is EBV (the cause of infectious mononucleosis, Burkitt's lymphoma and nasopharyngeal carcinoma) and the γ2 viruses include Kaposi's sarcoma herpesvirus (KSHV) and the important model virus MHV-68. Unique among the herpesviruses, γ-herpesviruses encode genes that are only expressed during latency. They also encode oncogenes and are associated with malignancy.
Herpesviruses are the most ancient known mammalian viruses and are extremely successful in evolutionary terms. Many family members infect a high percentage of individuals of their host species for the lifetime of the individual. Although the cell types they infect are diverse, herpesviruses share the ability to establish latent infection. True viral latency means that the virus can exist in host cells without reproducing itself, in contrast to persistence, when basal viral replication continues (the distinction is imperfect). Herpesviruses reactivate and replicate in a fully immune host, which enables their transmission to children, the next generation of hosts.
Herpesviruses live on the edge: they depend on immune control for their host's (and hence their own) survival, yet must impair immunity to avoid eradication. Severely immunocompromised patients often succumb to reactivated herpesvirus infections (especially CMV). The stability of the host-virus relationship shows that this is a robust and well-buffered equilibrium, only rarely does the host or the virus gain the advantage. This equilibrium is generally maintained only when herpesviruses infect their natural host. For example, the monkey virus herpes B virus causes an unapparent infection in monkeys but kills 70% of the people who are infected.
Dissecting the complex relationship between herpesviruses and the immune system depends on mouse models, where it is possible to genetically alter both virus and host. The best characterized mouse herpesviruses are MCMV, a pathogen that is ubiquitous in wild mice, and MHV-68, originally isolated from voles but nevertheless able to infect and establish latent infection in laboratory mice.
Immune control of MCMV is complex. Both type I IFN and IFN-γ and NK cells play major roles in containing primary infection32,33, yet acute infection is invariably fatal in the absence of B and T cell responses. Antibodies, NK cells, CD8+ T cells and CD4+ T cells all contribute to preventing dissemination of reactivated latent virus. Reactivation in antibody-deficient mice requires depleting any two of these three effector cell populations34. For the γ2 herpesvirus MHV-68 the situation is complicated further by a biphasic acute infection and limited understanding of the genetic program of latent infection. Again, all components of the immune response are involved in host defense35. After intranasal infection, MHV-68 replicates acutely in the lungs and disseminates to the spleen. This first phase of infection is controlled primarily by type I IFN, with some help from CD4+ and CD8+ T cells. A second (mononucleosis-like) phase of the infection ensues, characterized by a massive CD4+ T cell–dependent splenic B cell expansion and increase in viral load. Various viral transcripts can be detected, but infectious virus cannot be recovered, so virus at this stage is considered latent. This phase is gradually controlled by antibodies and both T cell subsets. Finally, a poorly characterized true latency is established, during which depleting both T cell subsets does not lead to virus reactivation.
Acute viruses: VIPR-free?
Viruses that are transmitted via acute infections are not known to encode VIPRs. This cannot be easily dismissed as evolutionary incompetence. HSV-encoded ICP47 is the “poster” protein for how easy blocking presentation can be. This highly effective VIPR comprises only 87 amino acids (even a 32-residue fragment is highly active). It would be trivial for even small viruses with severe nucleic acid packaging constraints to encode something similar (this could easily be accommodated in an overlapping reading frame, requiring no increase in genome size). This suggests that blocking antigen presentation to CD8+ T cells is useful only under a highly restricted set of circumstances. Perhaps blocking MHC class I expression greatly sensitizes cells for NK recognition, and the costs of countering both CD8+ T cells and NK cells are just too steep for acute viruses. Or perhaps very acute viruses are transmitted too rapidly for CD8+ T cells to have an impact on their evolution.
MHC class II VIPRs
Interference with the MHC class II antigen-processing pathway has been described for several herpesviruses36,37,38. As with cross-priming of CD8+ T cells, MHC class II VIPRs are not expected to block the presentation of exogenous antigens to CD4+ T cells, as—by definition—the APC is not infected and will not therefore express the VIPR. Thus, as with MHC class I VIPRs, class II VIPRs would have to act in vivo by interfering with CD4+ T cell–mediated clearance of virus-infected cells. For viruses whose transmission entails infection of MHC class II–expressing cells, this is certainly possible, but the physiological relevance of MHC class II VIPRs remains to be established.
Establishing VIPR function
Physiological relevance is, of course, the ultimate measure of VIPR function. Demonstrating that a viral protein can interfere with some aspect of antigen presentation or CD8+ T cell activation in vitro is not synonymous with equivalent in vivo function. There are examples of individual VIPRs that interfere with the function of numerous host proteins in cultured cells, and it is possible (even likely) that some of the interactions are not important (admittedly, disproving something is difficult). The degree of skepticism regarding the relevance of in vitro findings should reflect the deviation from natural circumstances, for example, when transfection is used to achieve expression that vastly exceeds that seen in infected cells39. Such approaches reveal potential functions, but eventually must be reinforced by evidence obtained from animal experiments.
We do not intend to demean the value of in vitro studies. Some VIPRs act with such efficiency in cultured cells (interference with TAP function by HSV ICP47 protein is the best example), as to make it exceedingly unlikely that the interaction is insignificant. Establishing VIPR function in vivo is never easy. For human viruses, often the best that can be achieved is demonstrating interference with CTL recognition of virus-infected cells in vitro. This has been shown for some VIPRs, notably HIV Nef40 and HCMV pp6530. With few exceptions, human viruses do not naturally infect other species, so it is simply not possible to rigorously study their VIPRs under natural circumstances. VIPRs can demonstrate similar interactions with homologous targets from different species: for example, AdE319K and HCMV US2 and US11 down-regulate mouse MHC class I, but HSV ICP47 does not. When a VIPR does function in mice, it may be possible to establish an in vivo function for a given gene product from a human virus, but again caution is necessary. No matter how the VIPR is expressed, the conditions of expression will differ from the natural situation in a manner that can profoundly affect its function. Variables include the amount of expression, cell type and a lack of other appropriate viral gene products. This basically limits thorough investigation of VIPRs to those expressed by natural animal pathogens. Given the vast superiority of mice as an experimental system, this puts the focus on mouse viruses, although the importance of the AIDS epidemic has made the rhesus macaque model of SIV an important exception.
The basic rules for establishing the in vivo function of VIPRs (and other immunomodulatory proteins) were spelled out in a report by Koszinowski and colleagues41 (extending a previously pioneered approach to demonstrating the function of a viral complement–interfering protein42). First, disabling the gene reduces the fitness of the mutant virus in vivo without affecting its ability to replicate in tissue culture. Ideally, fitness is defined by transmission. This is technically difficult to determine (for some viruses, well nigh impossible), so viral titers are taken as a surrogate. Second, reinserting the gene into the mutant virus (generating a “rescuant”) restores reproductive fitness. This step is needed to ensure that other alterations in the mutant virus inadvertently introduced during its generation are not responsible for the phenotype. Small viruses can be completely sequenced, but this remains impractical for herpesviruses and other large viruses. Third, the fitness of the mutant virus is restored by interfering with CD8+ T cell function, either with an appropriate genetically deficient mouse (TAP-deficient, for example) or by treating mice with a CD8+ T cell–depleting antibody.
Just as Koch's postulates (KPI) for demonstrating the cause of transmissible diseases cannot constitute proof (in the mathematical sense of the word) that a given agent is responsible for a given disease, fulfilling Koszinowski's postulates (KPII) does not guarantee that a gene product evolved to perform the proposed function. At the very least, the following caveats need to be considered.
Even the simple proposition that removing a gene should reduce fitness and reveal its function is problematical. If there is true redundancy in either the viral genes or the host defense mechanisms (or in both) such an effect may be difficult to demonstrate. Multifunctional viral genes (which are common) raise obvious problems. Here the researcher must attempt to make mutants in which the putative immune evasion phenotype is lost while other functions are maintained (this presumes that the other functions are known).
Excluding extraneous effects due to inadvertent genetic alterations is usually tidily dealt with by making resucants, but even this can be misinterpreted. If disruption of the targeted gene disrupts expression or control of an adjacent or unappreciated overlapping open-reading frame (ORF) that contributes to the phenotype, restoring the original genetic sequence will also restore the phenotype. Replacing the gene elsewhere in the genome is theoretically a better control, but rarely done. An alternative is to make multiple independent mutants, disrupting the gene in different ways (for example, mutating the promoter or the ORF) and showing that they have the same phenotype.
Equalizing the fitness of the mutant and wild-type virus by deleting the putatively targeted host mechanism (so called “genetic complementation”) is an intellectually appealing and essential part of KPII. Yet interpreting such experiments can be fraught with hazards. The first comes from making qualitative interpretations of an outcome that may result from an unrelated quantitative advantage. That is, if the balance between virus and host is such that CD8+ T cells achieve good but imperfect control, in theory, any (that is, VIPR-unrelated) reduction in viral fitness could tip the balance and enable CD8+ T cells to achieve complete control. Another problem is that genes may have unappreciated targets whose function contributes to the phenotype examined. The MCMV class I homolog m144 is assumed to act as a ligand for inhibitory NK receptors. Indeed, deleting m144 reduced virus fitness, the rescuant showed wild-type fitness and the mutant and wild-type fitnesses were equalized by NK cell depletion43. However, the appreciation that the ligand for the homologous HCMV class I protein UL18 is in fact a leukocyte Ig-like receptor (LIR) expressed more on macrophages than NK cells suggests other interpretations for the data. For example, m144 may inhibit MCMV-induced macrophage or DC activation, reducing cytokine secretion and hence the NK cell response. This scenario shows that our interpretation of KPII relies on current models through whose smoky glass we interpret the data.
Despite these caveats, KPII remains the essential means for addressing the in vivo function of viral gene products. We now have a handful of examples of its application. The first application of KPII to establish the function of a VIPR was the seminal study that showed MCMV lacking m152 replicated less efficiently than wild-type or rescuant virus 7 days after infection, a difference that was abolished by depleting CD8+ T cells41. Although welcome as the first demonstration of VIPR function in vivo, the real surprise of this article was just how modest the effect was (usually less than a tenfold difference). This raises an important question: do VIPRs generally exert such a modest effect? Or is there something that we are missing about m152? Indeed, m152 had an even more profound effect on the activity on NK cells and hence on viral titers at day 3, due to its inhibition of the NK activating ligand H-6044. So the “true” evolutionary function of m152 is difficult to establish. The most important message from m152 may be that evolution favors complexity and that we need to interpret experiments with this in mind.
MHV-68 lacking the M3 chemokine-binding protein replicated acutely in lung epithelium and initially seeded the spleen to almost the same extent as wild-type virus. However, in contrast to wild-type virus, the mutant virus did not cause B cell expansion in the spleen with its concomitant amplification of latent viral load unless CD8+ T cells were depleted18. The interpretation favored by the authors—that M3 prevents chemokine-driven CD8+ T cells trafficking and impairs their ability to control latent virus—is reasonable. The difficulty in interpreting this type of experiment (see above) is underscored by the fact that an independent M3 virus carrying a deletion had little, if any, latency deficit45.
MHV-68 K3 interferes directly with CD8+ T cell recognition of infected cells46. The K3-deficient mutant, like the M3-deficient mutant, was primarily impaired during the second mononucleosis-like phase of infection, which was attributed to more efficient CD8+ T cell function47. These two mutants suggest CD8+ T cells must be disabled for splenic amplification of the latent virus pool and, therefore, that MHV-68 VIPRs are important for this phase. It is not known how closely infection of mice mimics infection of the natural host (voles); consequently, the real contribution made by VIPRs to the fitness (that is, transmission) of γ2 herpesviruses may be difficult to determine in this model.
The role of MHC class I down-regulation by Nef has been investigated in the SIV model48. Three rhesus macaques were infected with SIV with a Nef that was modified (Y223F) to disable its VIPR activity without affecting its myriad other functions. Four weeks after infection, the majority of viruses recovered from each animal had reverted to wild-type Nef. Although this suggests that MHC class I down-regulation has a strong influence on viral fitness, further evidence is needed to fulfill KPII. Does reversion occur in CD8+ T cell–depleted animals? Is SIV replication compromised by the introduction of a mutation that is more difficult to revert?
These examples represent the vanguard of what we hope will be a serious long-term effort to assess the in vivo function of VIPRs and other immunomodulatory proteins. Although the complexity of the immune system makes such studies difficult, they should provide a bounty of findings that will enliven the enterprise and deepen our insight into the general workings of the immune system.
Escape mutants
Viruses have another potential means for avoiding recognition by CD8+ T cells. RNA viruses have extremely high mutation rates, which means that they exist in nature as a diverse genetic mix. Of course, most mutations are harmful, and in the absence of selective pressure the average genetic composition of the population tends to remain fairly stable, reflecting the optimal gene function. However, these viruses have the potential to rapidly decrease the immunogenicity of their gene products. This is not terribly difficult due to the number of filters in place that limit the potential immunogenicity of peptides. Consequently, the chance that any given peptide in a protein will be immunogenic with any given MHC class I allomorph is ∼1/200011.
Highly artificial model systems have shown that CD8+ T cells can select for antigenic escape mutants in mice. Clear evidence has been presented for the selection of escape variants in SIV and HIV infections49,50,51. Individual escape mutants may enable a virus to persist in one host and hence improve transmission to others. MHC polymorphism, however, limits the advantage in a new host, which is likely to possess an MHC allomorph that binds to different peptides. Thus, RNA viruses profit from their ability to rapidly generate new variants per se, rather than generate any given escape variant. Indeed, preventing escape variants is thought to be the primary evolutionary selection factor for MHC polymorphism (ask any viral immunologist).
In theory, viruses could avoid MHC polymorphism by creating proteins that are antigenically identical to the host. This would enable viral avoidance of antibody recognition as well. Despite the high hopes of immunologists, if viruses exploit mimicry, they do so subtly and infrequently; viral proteins are usually only distantly related to host proteins, even those with similar functions.
Horizons
We will end our review by considering what we have learned from our fascination with VIPRs and what might be gleaned from further studies. Following the long tradition of viral gene products, VIPRs have contributed to our understanding of basic cellular biological processes. An early contribution came from the first-defined VIPR, AdE3-19K, which led to the identification of ER-retention motifs in the cytosolic domains of membrane proteins52. More recently, HCMV US2 and US11 greatly facilitated studies in mammalian cells of retro-translocation of ER proteins into the cytosol.
Overall, however, our understanding of the classical MHC class I pathway has enabled us to understand the mechanistic actions of VIPRs rather than vice versa. VIPRs may yet teach us something about antigen presentation. Two classes of VIPRs (HCMV US1153 and the K3 from KSHV and MHV-6854) ubiquitylate MHC class I heavy chain cytosolic domains to target it for destruction, either in the ER or a post-medial Golgi complex compartment. The lysine ubiquitylated through the action of K3 is highly conserved among MHC class I allomorphs, which suggests that this process has a normal counterpart, perhaps identifying MHC class I molecules lacking peptides for internalization and degradation. No doubt this will soon be sorted out. VIPRs may also fulfill their promise and become powerful tools for sorting out the contributions of direct and cross-presentation in activating naïve virus-specific CD8+ T cells28.
Occurring simultaneously with a great leap in deciphering the molecular basis for NK cell recognition, the study of herpesvirus immune evasion has played an important role in the expansion of our concept of the extended family of MHC class I molecules and their cognate receptors. It is likely that when the dust settles, the very definition of the MHC class I family and its role in immunobiology will have to be reconsidered. The HCMV MHC class I homolog UL18 seemed tailor made as an NK cell decoy, but identification of a cellular ligand led to the discovery of an entire new family of killer cell inhibitory receptor (KIR)-like genes, the LIRs55. The LIRs also recognize classical MHC class I gene products, but are expressed more by macrophages and B cells than by NK cells or T cells. The functions of the HCMV LIR ligand56 and the LIRs expressed by myeloid cells are a complete mystery.
Meanwhile, NK cell biologists have extended the concept of “class I–like” by identifying ligands for the activating NK receptor NKG2D. An HCMV gene, UL16, played an important role in this story57. Although homology of some of these ligands to class I molecules is so low as to strain credulity, it seems sufficient for the MCMV VIPR m152, which targets both classical MHC class I and a gene product that hardly looks like one (H-60)44. Extending the boundaries still further, the MHC class I family has recently accepted another cohort of relatives, with the identification of another MCMV gene, m157, as the ligand for the activating NK receptor Ly49H58,59. m157 has distant secondary structural homology to MHC class I, as do a handful of other MCMV genes, including m15259 (and L. Lanier, personal communication). This story remains to be written, but it is clear that VIPRs are providing crucial insights into both the identities and functions of members the far-flung MHC class I family.
With the possible exception of MHV-68 K3, we lack a clear demonstration that VIPRs are necessary for viral persistence via interfering with CD8+ T cell recognition of infected cells. The odds still favor the possibility that this property is a selective factor driving VIPR evolution. However, our broadened awareness of the biology of the extended MHC class I family and its ligands forces us to consider broader hypotheses for the interest of viruses in MHC class I molecules. It would be an ironic twist in the VIPR story if interference with antigen presentation to CD8+ T cells turns out to be only a minor part of their complete job description. After all, snakes enjoy a varied diet.
References
Blanden, R.V. Mechanisms of recovery from a generalized viral infection: mousepox. I. The effects of anti-thymocyte serum. J. Exp. Med. 132, 1035–1054 (1970).
Blanden, R.V. Mechanisms of recovery from a generalized viral infection: mousepox. II. Passive transfer of recovery mechanisms with immune lymphoid cells. J. Exp. Med. 133, 1074–1089 (1971).
Blanden, R.V. Mechanisms of recovery from a generalized viral infection: mousepox. 3. Regression infectious foci. J. Exp. Med. 133, 1090–1104 (1971).
Walter, E.A. et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N. Eng. J. Med. 333, 1038–1044 (1995).
Brodie, S.J. et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nature Med. 5, 34–41 (1999).
Khanna, R. et al. Activation and adoptive transfer of Epstein-Barr virus-specific cytotoxic T cells in solid organ transplant patients with post-transplant lymphoproliferative disease. Proc. Natl. Acad. Sci. USA 96, 10391–10396 (1999).
Gadola, S.D., Moins-Teisserenc, H.T., Trowsdale, J., Gross, W.L. & Cerundolo, V. TAP deficiency syndrome. Clin. Exp. Immunol. 121, 173–178 (2000).
Rock, K.L. & Goldberg, A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 17, 739–779 (1999).
Pamer, E. & Cresswell, P. Mechanisms of MHC class I–restricted antigen processing. Annu. Rev. Immunol. 16, 323–358 (1998).
Yewdell, J.W., Schubert, U. & Bennink, J.R. At the crossroads of cell biology and immunology: DRiPs and other sources of peptide ligands for MHC class I molecules. J. Cell Sci. 114, 845–851 (2001).
Yewdell, J.W. & Bennink, J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 17, 51–88 (1999).
Posavad, C.M., Newton, J.J. & Rosenthal, K.L. Infection and inhibition of human cytotoxic T lymphocytes by herpes simplex virus. J. Virol. 68, 4072–4074 (1994).
Andrews, D.M., Andoniou, C.E., Granucci, F., Ricciardi-Castagnoli, P. & Degli-Esposti, M.A. Infection of dendritic cells by murine cytomegalovirus induces functional paralysis. Nature Immunol. 2, 1077–1084 (2001).
Moutaftsi, M., Mehl, A.M., Borysiewicz, L.K. & Tabi, Z. Human cytomegalovirus inhibits maturation and impairs function of monocyte-derived dendritic cells. Blood 99, 2913–2921 (2002).
Engelmayer, J. et al. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J. Immunol. 163, 6762–6768 (1999).
Salio, M., Cella, M., Suter, M. & Lanzavecchia, A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 29, 3245–3253 (1999).
Norbury, C.C., Malide, D., Gibbs, J.S., Bennink, J.R. & Yewdell, J.W. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nature Immunol. 3, 265–271 (2002).
Bridgeman, A., Stevenson, P.G., Simas, J.P. & Efstathiou, S. A secreted chemokine binding protein encoded by murine γ herpesvirus-68 is necessary for the establishment of a normal latent load. J. Exp. Med. 194, 301–312 (2001).
Tortorella, D., Gewurz, B.E., Furman, M.H., Schust, D.J. & Ploegh, H.L. Viral subversion of the immune system. Annu. Rev. Immunol. 18, 861–926 (2000).
Alcami, A. & Koszinowski, U.H. Viral mechanisms of immune evasion. Immunol. Today 21, 447–55 (2000).
Yewdell, J.W. & Bennink, J.R. Mechanisms of viral interference with MHC class I antigen processing and presentation. Annu. Rev. Cell Dev. Bio. 15, 579–606 (1999).
Bevan, M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 143, 1283–1288 (1976).
Levitskaya, J. et al. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 375, 685–688 (1995).
Blake, N. et al. Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity 7, 791–802 (1997).
Sigal, L.J. & Rock, K.L. Bone marrow-derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and - independent pathways of antigen presentation. J. Exp. Med. 192, 1143–1150 (2000).
Prasad, S.A., Norbury, C.C., Chen, W., Bennink, J.R. & Yewdell, J.W. Cutting edge: recombinant adenoviruses induce CD8 T cell responses to an inserted protein whose expression is limited to nonimmune cells. J. Immunol. 166, 4809–4812 (2001).
Norbury, C.C. et al. Multiple antigen-specific processing pathways for activating naive CD8+ T cells in vivo. J. Immunol. 166, 4355–4362 (2001).
Basta, S., Chen, W., Bennink, J.R. & Yewdell, J.W. Inhibitory effects of cytomegalovirus proteins US2 and US11 point to contributions from direct priming and cross-priming in induction of vaccinia virus-specific CD8+ T cells. J. Immunol. 168, 5403–5408 (2002).
Gold, M.C. et al. The murine cytomegalovirus immunodomulatory gene m152 prevents recognition of infected cells by M45-specific CTL, but does not alter the immunodominance of the M45-specific CD8 T cell response in vivo. J. Immunol. 169, 359–365 (2002).
Gilbert, M.J., Riddell, S.R., Plachter, B. & Greenberg, P.D. Cytomegalovirus selectively blocks antigen processing and presentation of its immediate-early gene product. Nature 383, 720–722 (1996).
Reimann, J. & Schirmbeck, R. Alternative pathways for processing exogenous and endogenous antigens that can generate peptides for MHC class I-restricted presentation. Immunol. Rev. 172, 131–152 (1999).
Orange, J.S. & Biron, C.A. Characterization of early IL-12, IFN-αβ, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J. Immunol. 156, 4746–4756 (1996).
Orange, J.S. & Biron, C.A. An absolute and restricted requirement for IL-12 in natural killer cell IFN-γ production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J. Immunol. 156, 1138–1142 (1996).
Polic, B. et al. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J. Exp. Med. 188, 1047–1054 (1998).
Doherty, P.C., Christensen, J.P., Belz, G.T., Stevenson, P.G. & Sangster, M.Y. Dissecting the host response to a γ-herpesvirus. Phil. Trans. R. Soc. Lond. B 356, 581–593 (2001).
Redpath, S., Angulo, A., Gascoigne, N.R. & Ghazal, P. Murine cytomegalovirus infection down-regulates MHC class II expression on macrophages by induction of IL-10. J. Immunol. 162, 6701–6707 (1999).
Tomazin, R. et al. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nature Med. 5, 1039–1043 (1999).
Cebulla, C.M. et al. Human cytomegalovirus disrupts constitutive MHC class II expression. J. Immunol. 169, 167–176 (2002).
Liu, X., Schrager, J.A., Lange, G.D. & Marsh, J.W. HIV Nef-mediated cellular phenotypes are differentially expressed as a function of intracellular Nef concentrations. J. Biol. Chem. 276, 32763–32770 (2001).
Collins, K.L., Chen, B.K., Kalams, S.A., Walker, B.D. & Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401 (1998).
Krmpotic, A. et al. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 190, 1285–1296 (1999).
Lubinski, J.M. et al. Herpes simplex virus type 1 glycoprotein gC mediates immune evasion in vivo. J. Virol. 72, 8257–8263 (1998).
Farrell, H.E. et al. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature 386, 510–514 (1997).
Krmpotic, A. et al. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nature Immunol. 6, 529–535 (2002).
van Berkl, V. et al. Critical role for a high-affinity chemokine-binding protein in γ-herpesvirus-induced lethal meningitis. J. Clin. Invest. 109, 905–914 (2002).
Stevenson, P.G., Efstathiou, S., Doherty, P.C. & Lehner, P.J. Inhibition of MHC class I-restricted antigen presentation by γ 2-herpesviruses. Proc. Natl. Acad. Sci. USA 97, 8455–8460 (2000).
Stevenson, P.G. et al. K3-mediated evasion of CD8+ T cells aids amplification of a latent γ-herpesvirus. Nature Immunol. 3, 733–740 (2002).
Munch, J., Stolte, N., Fuchs, D., Stahl-Hennig, C. & Kirchhoff, F. Efficient MHC class I major histocompatibility complex down-regulation by simian immunodeficiency virus Nef is associated with a strong selective advantage in infected rhesus macaques. J. Virol. 75, 10532–10536 (2001).
Allen, T.M. et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407, 386–390 (2000).
Moore, C. et al. Evidence of HIV adaptation to HLA-restricted immune responses at a population level. Science 296, 1439–1443 (2002).
Goulder, P.J. et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412, 334–338 (2001).
Paabo, S., Bhat, B.M., Wold, W.S.M. & Peterson, P.A. A short sequence in the COOH-terminus makes an adenovirus membrane glycoprotein a resident of the endoplasmic reticulum. Cell 50, 311–317 (1987).
Shamu, C.E., Story, C.M., Rapoport, T.A. & Ploegh, H.L. The pathway of US11-dependent degradation of MHC class I heavy chains involves a ubiquitin-conjugated intermediate. J. Cell Biol. 147, 45–58 (1999).
Hewitt, E.W. et al. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J. 21, 2418–2429 (2002).
Cosman, D. et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 7, 273–282 (1997).
Cosman, D., Fanger, N. & Borges, L. Human cytomegalovirus, MHC class I and inhibitory signalling receptors: more questions than answers. Immunol. Rev. 168, 177–185 (1999).
Cosman, D. et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14, 123–133 (2001).
Arase, H., Mocarski, E.S., Campbell, A.E., Hill, A.B. & Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326 (2002).
Smith, H. et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 19, 8826–8831 (2002).
Levitskaya, J., Sharipo, A., Leonchiks, A., Ciechanover, A. & Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 94, 12616–12621 (1997).
Hill, A. et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature 375, 411–415 (1995).
Fruh, K. et al. A viral inhibitor of peptide transporters for antigen presentation. Nature 375, 415–418 (1995).
Hinkley, S., Hill, A.B. & Srikumaran, S. Bovine herpesvirus-1 infection affects the peptide transport activity in bovine cells. Virus Res. 53, 91–96 (1998).
Ahn, K. et al. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 6, 613–621 (1997).
Bennett, E.M., Bennink, J.R., Yewdell, J.W. & Brodsky, F.M. Cutting edge: adenovirus E19 has two mechanisms for affecting class I MHC expression. J. Immunol. 162, 5049–5052 (1999).
Gruhler, A., Peterson, P.A. & Fruh, K. Human cytomegalovirus immediate early glycoprotein US3 retains MHC class I molecules by transient association. Traffic 1, 318–325 (2000).
Jones, T.R. et al. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc. Natl. Acad. Sci. USA 93, 11327–11333 (1996).
Kavanagh, D.G., Koszinowski, U.H. & Hill, A.B. The murine cytomegalovirus immune evasion protein m4/gp34 forms biochemically distinct complexes with class I MHC at the cell surface and in a pre-golgi compartment. J. Immunol. 167, 3894–3902 (2001).
Wiertz, E.J.H. et al. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769–779 (1996).
Wiertz, E.J.H.J. et al. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384, 432–438 (1996).
Boname, J.M. & Stevenson, P.G. MHC class I ubiquitination by a viral PHD/LAP finger protein. Immunity 15, 627–636 (2001).
Kerkau, T. et al. The human immunodeficiency virus type-1 (HIV-1) Vpu protein interferes with an early step in the biosynthesis of MHC class I molecules. J. Exp. Med. 185, 1295–1305 (1997).
Schubert, U. et al. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 72, 2280–2288 (1998).
Ziegler, H. et al. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 6, 57–66 (1997).
Kavanagh, D.G., Gold, M.C., Wagner, M., Koszinowski, U.H. & Hill, A.B. The Multiple immune-evasion genes of murine cytomegalovirus are not redundant. M4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J. Exp. Med. 194, 967–978 (2001).
Ziegler, H., Muranyi, W., Burgert, H.G., Kremmer, E. & Koszinowski, U.H. The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 19, 870–881 (2000).
Reusch, U. et al. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18, 1081–1091 (1999).
Piguet, V. et al. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nature Cell Biol. 2, 163–167 (2000).
Ishido, S. et al. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity 13, 365–374 (2000).
Kleijnen, M.F. et al. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER, which is not retained but is transported to the cell surface. EMBO J. 16, 685–694 (1997).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yewdell, J., Hill, A. Viral interference with antigen presentation. Nat Immunol 3, 1019–1025 (2002). https://doi.org/10.1038/ni1102-1019
Issue Date:
DOI: https://doi.org/10.1038/ni1102-1019
This article is cited by
-
Transcriptome of nasopharyngeal samples from COVID-19 patients and a comparative analysis with other SARS-CoV-2 infection models reveal disparate host responses against SARS-CoV-2
Journal of Translational Medicine (2021)
-
Screening HLA-A-restricted T cell epitopes of SARS-CoV-2 and the induction of CD8+ T cell responses in HLA-A transgenic mice
Cellular & Molecular Immunology (2021)
-
Role of HTLV-1 orf-I encoded proteins in viral transmission and persistence
Retrovirology (2019)
-
Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes
Nature Communications (2019)
-
Immune selection during tumor checkpoint inhibition therapy paves way for NK-cell “missing self” recognition
Immunogenetics (2017)
