
Abstract
Viruses have evolved mechanisms to avoid the host immune system, including means of escaping detection by both the innate and adaptive immune responses. Natural killer (NK) cells are a central component of the innate immune system and are crucial in defense against certain viruses. To attain a state of chronic infection, some successful viruses have developed specific mechanisms to evade detection by and activation of NK cells. These NK cell–specific evasion mechanisms fall into distinct mechanistic categories used in numerous virus families.
Similar content being viewed by others
Main
NK cells are lymphocytes that do not undergo genetic recombination events to increase their affinity for particular ligands, and are thus considered part of the innate immune system. They are capable of mediating cytotoxic activity and of producing cytokines after ligation of a variety of germline-encoded receptors. NK cells mediate direct lysis of target cells by releasing cytotoxic granules containing perforin and granzymes, or by binding to apoptosis-inducing receptors on the target cell. They also secrete cytokines such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) during infection and inflammation. Several receptors that can activate NK cells have been identified, including the human natural cytotoxicity receptors NKp30, NKp44, NKp461 and Ly49D and Ly49H in the mouse2. Although the specificities of many NK cell–activating receptors are still unknown, some recognize viral products; these include influenza hemagglutinin, recognized by NKp463, and murine cytomegalovirus (MCMV) m157, recognized by Ly49H4,5. Other well known molecules can also function as activation receptors in NK cells, including leukocyte function–associated antigen 1 (LFA-1)6 and the CD2 family7. NK cell responses are also coordinated and modulated by cytokines, including IFN-α, IFN-β, interleukin 2 (IL-2), IL-12, IL-15 and IL-188.
Because of the possible consequences of NK cell activation, normal host cells must be able to readily and effectively inhibit NK cells. Various inhibitory receptors are consistently expressed by subsets of NK cells, including killer-cell immunoglobulin-like receptors (KIR), immunoglobulin-like inhibitory receptors (ILT) and the lectin-like heterodimer CD94-NKG2A1. These receptors bind to host MHC class I molecules and transmit inhibitory signals to the NK cell through intracellular tyrosine-based inhibitory motifs (ITIMs) contained in their cytoplasmic domains. Signaling via coreceptors with inhibitory potential, such as CD81, may also silence NK cells. Thus, NK cells can mediate powerful effector functions, but are effectively regulated by healthy host cells under normal circumstances.
NK cells are activated during a wide variety of viral infections by virus-induced type I IFNs8. Studies in animal models, however, have demonstrated that NK cells are required for clearance of only certain viruses, including herpesviruses. The importance of NK cell defense against these viruses is highlighted by the susceptibility of mice depleted of NK cells to experimental infection and by the invasive or disseminated viral disease that is associated with naturally occurring NK cell deficiencies in humans8,9,10,11. Many of these pathogens have effective means of avoiding the adaptive immune response. In eluding T cells, however, these viruses might have increased their susceptibility to NK cell–mediated defenses.
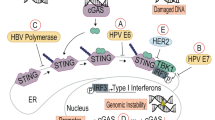
Members of the herpesvirus, papillomavirus, retrovirus, poxvirus and flavivirus families have developed mechanisms to evade the NK cell response. These fall into five categories (Fig. 1): expression of virally encoded MHC class I homologs; selective modulation of MHC class I protein expression by viral proteins; virus-mediated inhibition of activating receptor function; production of virally encoded cytokine-binding proteins or cytokine-receptor antagonists; and direct viral effects on NK cells. The putative purpose of these mechanisms is to block NK cell activity. Examples of each strategy (summarized in Table 1) are discussed below. Many other viruses probably use these strategies, and additional viral products may affect NK cell functions. This discussion is limited to viral infections and viral gene products known to affect the NK cell response.

The strategies by which viruses evade NK cells fall into five categories and are depicted in the interaction between a virus infected target cell (left) and an NK cell (right). (a) NK cells can be inhibited by a viral MHC class I homolog with structural similarity to endogenous host class I that binds to inhibitory class I receptors on NK cells. (b) Viruses can inhibit expression of HLA-A and HLA-B, resulting in a relative increase in HLA-C and HLA-E on the surface of the target cell; these inhibit NK cells through the class I inhibitory receptors CD94-NKG2A and KIR, respectively. Alternatively, viral gene expression can result in selectively increased expression of HLA-E, which inhibits NK cells through CD94-NKG2A. (c) Virus-encoded proteins can function as cytokine binding proteins that block the action of NK cell activating cytokines. In addition, viruses can produce homologs, or increase host production of cytokines that inhibit NK cells. (d) NK cell activities can also be avoided by decreased expression of NK cell–activating ligands in virus-infected target cells, which prevent signal transduction via NK cell–activating receptors. To achieve the same end, viruses can encode antagonists of the activating receptor–ligand interaction. (e) Viruses can also directly inhibit NK cells by infecting them or using envelope proteins to ligate NK cell inhibitory receptors. Proteins outlined in red are virally encoded. Each mechanism corresponds to the similarly numbered section of the text where additional details and examples are provided.
Inhibition of NK cells by viral homologs of MHC class I
Virus-encoded homologs of cellular MHC class I genes represent the earliest recognized viral mechanism for evading NK cells. Many viruses evade T cell recognition by down-regulating class I molecules on the surface of the host cell (discussed below). In theory this leaves infected cells susceptible to NK lysis owing to the reduced opportunity for class I molecules—human leukocyte antigen C (HLA-C) and HLA-E—to engage NK cell inhibitory receptors. The discovery of a class I homolog, UL18, in the genome of human cytomegalovirus (HCMV)12 has led to speculation that these viral proteins might serve as NK cell decoys and ligate inhibitory receptors to block NK cell cytotoxicity in the absence of host class I molecules.
Early studies in which HCMV UL18 was expressed in the HLA-A, HLA-B and HLA-C–deficient B cell line 721.221 demonstrated a CD94-dependent inhibition of killing by various NK cell lines and clones13. Subsequently, a principal ligand of the CD94-NKG2A inhibitory receptor was found to be HLA-E, a class I protein that is expressed only after forming a complex with a peptide nonamer derived mainly from the signal peptides of some of the class I proteins14. Further efforts to identify a nonamer in the UL18 sequence that would form a complex with HLA-E have been unsuccessful15. Thus, the mechanism underlying the reported inhibition of lysis of the human B cell line transfected with UL18 remains unexplained. It was not due to inadvertent selection of HLA-E–expressing 721.221 cells because immunoprecipitation of class I molecules from the 721.221-UL18 transfectants yielded a heavily glycosylated α-chain of the size of UL18 (∼66 kD) and not HLA-E (44 kD)13. In a contrasting report, however, UL18-transfected fibroblasts showed increased susceptibility to lysis by NK cell lines, and fibroblasts infected with wild-type HCMV were lysed more efficiently than those infected with a UL18-deficient HCMV16. Alternatively, the inhibitory activity mediated by UL18 may be due to its binding of ILT-2 (LIR-1), an inhibitory receptor expressed by B cells, monocytes and a subset of NK cells17. Thus, the role of UL18 in evasion of NK cells is not completely understood, but it may be an important mechanism by which certain subpopulations of NK cells are inhibited.
Compelling data support the notion that the MCMV MHC class I homolog m144 is involved in the inhibition of NK cells. An m144 deletion strain of MCMV is less virulent than the wild-type virus, and this effect is reversed after in vivo depletion of NK cells18. Transfection of m144 into the Raji cell line results in partial inhibition of antibody-dependent cellular cytotoxicity19. In addition, m144-transfected RMA-S lymphoma cells injected into mice are tumorigenic, whereas untransfected RMA-S tumors are rejected by NK cells20. Further investigation of m144 will benefit from identification of the cognate inhibitory receptor. The structures of m144 and UL18 are considerably different (for example, the latter binds peptides, the former does not), so that their receptors, mechanisms and functions may be distinct.
A newly described viral class I homolog, MCMV m157, has both activating and inhibitory effects upon NK cells, depending on the mouse strain from which the cells were derived. MCMV m157 is a ligand for the NK-activating receptor Ly49H in MCMV-resistant C57BL/6 mice and is predicted to share sequence and structural similarity with other nonclassical MHC class I genes4,5. Because a virus-encoded ligand that activates NK cells seems teleologically unsound from the standpoint of the virus, it is notable that m157 binds the putative inhibitory receptor Ly49I on a subset of NK cells in CMV-susceptible 129/J mice4. The significance of m157-induced activating and inhibitory functions is unknown, particularly in noninbred mice, but their existence suggests constant evolution in the host as well as the virus.
The dual specificity of MCMV m157 for NK-activating and NK-inhibitory receptors could provide new insight about HCMV UL18. Like m157, UL18 might exert different effects upon NK cells by interacting with distinct receptors in different in vitro or in vivo systems. In addition, the example of m157 is a useful reminder that viral homologs of MHC class I function in a complex, dynamic environment of viral and immune elements during in vivo infection.
Additional MHC class I homologs await characterization. Rat cytomegalovirus (RCMV) r144 has not been assayed for direct effects upon NK cell cytotoxicity, but lower virus titers occur in the spleens of rats infected with an r144-deleted strain of RCMV than in those infected with wild-type RCMV21. This suggests that like other class I homologs, r144 may be a ligand for an NK cell inhibitory receptor. Molluscum contagiosum virus (MCV) also encodes a class I homolog, MC080R, which is retained in the endoplasmic reticulum and Golgi of transfected cells22. Its effects on NK cell activity are presently unknown. Most promising is the recent identification of ten class I homologs in the MCMV genome, in addition to m144 and m1575. These genes were not detected earlier because they have structural rather than sequence similarity to known class I molecules. Their discovery is suggestive of the possibility that additional, undetected class I homologs might exist in other viral genomes as well.
Selective modulation of MHC class I allele expression
A common feature of many viral infections is the virus-induced modulation of class I expression. Viruses down-modulate class I molecules that are efficient at presenting viral peptides to CD8+ cytotoxic T cells (CTLs), such as HLA-A and HLA-B, to evade CTL-mediated destruction. In contrast, either HLA-C and HLA-E, the dominant ligands for NK cell–inhibitory receptors, are spared from virus-induced clearance from the cell surface or their expression is specifically enhanced. The selectivity of viral proteins for certain class I molecules appears to be indicative of a compromise ensuring that class I expression is diminished only to an extent that will still allow efficient inhibition of NK cells. Such a strategy appears to allow viruses to walk a fine line between the adaptive and innate arms of the immune system. A large number of viral proteins participate in class I down-regulation23. Here, the discussion will be limited to viral down-modulation of class I molecules as it relates to NK cell responses.
Many viral proteins cause class I molecules to deviate from their normal progression from the endoplasmic reticulum to the cell surface. At least four HCMV proteins, US2, US3, US6 and US11, function in this manner. US2 and US11 show a certain degree of selectivity in their targeting of class I molecules24,25. In particular, two dominant inhibitory receptor ligands, HLA-C and HLA-E, are resistant to either US2- or US11-mediated degradation, suggesting that virus-infected cells evade NK cell activity by sparing the class I molecules least effective at presenting viral peptides to CTL but most effective at inhibiting NK cells26,27. In contrast, class I molecules are nonselectively down-modulated from the cell surface by US3 and US6, and these proteins can partially inhibit the cell surface expression of HLA-C and HLA-G28. It is not clear whether the extent of down-modulation mediated by US3 and US6 is sufficient to make infected cells susceptible to NK cell lysis. Notably, none of these US proteins that down-regulate HLA expression inhibit the expression of the viral encoded class I homolog UL18, further demonstrating a selective targeting for NK cell evasion29.
MCMV possesses three genes, m04, m06 and m152, that selectively modulate class I expression; these genes are not homologous to the HCMV US genes and use slightly different modulation mechanisms. This indicates that HCMV and MCMV independently evolved systems to regulate class I expression. Notably, although the m04–class I protein complex inhibits CTLs, it is expressed on the cell surface where it could potentially inhibit NK cells30.
A second mechanism whereby certain viruses modulate class I expression is by accelerating the endocytosis of class I molecules from the cell surface. The nef gene, present in primate lentiviruses such as HIV-1, HIV-2 and SIV, encodes a 27-kD protein that is expressed early after infection and selectively down-modulates the expression of HLA-A and HLA-B, but not HLA-C or HLA-E31,32. Thus, HIV-infected target cells remain resistant to lysis by NK cells, and their resistance depends on the failure of Nef to down-modulate HLA-C and HLA-E from the cell surface. Nef acts by inducing the clathrin adaptor protein complex to recognize a tyrosine-based sorting motif in the cytoplasmic tail of HLA-A and HLA-B32. The resistance of HLA-C to Nef-induced down-regulation is due to locus-specific tyrosine-to-cysteine and aspartic-acid-to-asparagine substitutions in the cytoplasmic tail31,32. The SIV Nef protein also down-modulates monkey class I proteins via endocytosis but uses a COOH-terminal region of SIV Nef that is not present in HIV Nef, suggesting once again that viruses have repeatedly evolved multiple mechanisms to modulate class I expression33.
Acceleration of class I molecule endocytosis is also a feature of Kaposi's sarcoma–associated herpesvirus (KSHV). Two KSHV proteins, K3 and K5, induce the rapid endocytosis of class I proteins from the cell surface34,35. Whereas K3 down-modulates HLA-A, HLA-B, HLA-C and HLA-E, K5 is more selective and down-modulates only HLA-A and HLA-B36. Because K5 has other functions, it is difficult to evaluate the importance of HLA-C and HLA-E resistance to K5 endocytosis with regard to NK cells36.
In addition to their specific down-modulation of class I alleles, viruses also up-regulate certain class I alleles to evade NK cells. Previous work suggests that HCMV actively enhances the expression of HLA-E, the ligand for the inhibitory CD94–NKG2A receptor complex15,37. Cell surface expression of HLA-E requires binding of a nonamer peptide derived from the signal sequence of most HLA molecules38. The HCMV UL40 protein possesses a nonamer peptide homologous to HLA signal sequences and thus can enhance cell surface expression of HLA-E15,37. HLA-E binds the UL40 peptide in a TAP-independent manner, presumably bypassing the inhibitory effects of the HCMV US6 protein. In HCMV-infected cells, UL40 is necessary to mediate resistance to NK cell lysis in a CD94- and MHC class I–dependent manner39.
Additional investigation is required to test the effects of virus-induced MHC modulation on NK cell resistance. One complicating feature is that the retention of HLA-C on the cell surface may actually result in lysis of the virus-infected cells by NK cells possessing the cognate activating KIR but no inhibitory KIR40,41. Similarly, because HLA-E also binds to the CD94-NKG2C–activating receptor14, UL40-induced up-regulation of HLA-E could potentially activate NK cells. Thus, it is likely that selective HLA regulation contributes to viral evasion of NK cells, but the relative importance of this mechanism is uncertain.
Virus-mediated inhibition of activating receptor function
In addition to receptors with potent inhibitory capabilities, NK cells have receptors whose ligation can induce cytotoxicity, proliferation and cytokine production1. Several such activating receptors have been characterized, and some recognize putative ligands that include specific viral products3,4,5. An effective viral evasion strategy, therefore, would be to interfere with the process of activating receptor ligation. Several means to this end have been identified.
The most commonly documented mechanism of interference with activating receptor function is virus-mediated down-regulation of activating receptor ligands in infected cells. For example, certain strains of HCMV increase the resistance of their infected host cells to NK cell cytotoxicity by down-regulating LFA-342. This regulation, which is independent of the virus effects on class I molecules discussed previously, presumably interferes with the binding of LFA-3 to the NK cell–activating receptor, CD2. In addition, MCMV m152 encodes gp40, a protein that presumably inhibits surface expression of the murine ligand for the NKG2D-activating receptor, H-6043. Cells infected with m152-deleted MCMV have greater expression of H-60 and are more readily lysed by NK cells. An additional example of host-cell ligand regulation as an evasion strategy is found in KSHV. Transfection of target cells with the K5 gene of KSHV results in decreased expression of ICAM-1 and B7-2, both of which can serve as ligands for NK cell–activating receptors36,44. The K5-mediated reduction in the surface expression of these ligands results in target-cell escape from NK cell cytotoxicity in certain in vitro NK cell systems36, but not in others44. K5 is an E2 ubiquitin ligase and thus probably directs the ubiquitination of the NK cell–activating receptor ligands it targets, ultimately resulting in their degradation34. As a group, the host-cell molecules targeted by viruses have functions other than as NK cell–activating receptor ligands and thus these examples, although relevant, are not entirely specific to NK cells. The discovery of ligands for activating receptors specific to NK cells will probably lead to fruitful studies of viral regulation of their expression.
A second way that viruses may interfere with activating receptor function is by virus-induced modification of the ligand on target-cell surfaces. Infection of certain target cells by HIV, human T cell lymphotrophic virus I (HTLV-I) or HTLV-II can result in resistance to NK cell lysis associated with sialylation of surface molecules45. Although target-cell binding is not impaired by infection, chemical removal of sialic acid restores susceptibility to NK cell cytotoxicity. These receptor modifications have not been specifically seen on known NK cell ligands, but it can be reasoned that activating receptor ligands would be the most likely targets of virus-induced sialylation. It is to be hoped that follow-up studies will reveal the viral proteins responsible for, and specific cellular targets of, this evasion strategy.
A third mechanism for avoiding NK cell activation is antagonism of the activating receptor and ligand interaction. An example is the interaction of an HCMV encoded protein with the NKG2D receptor. NKG2D is an activating receptor expressed on NK cells and subsets of T cells that binds to the UL16-binding protein (ULBP) family of glycosylphosphatidylinositol-linked receptors and MHC class I–related molecules (MIC) in humans, as well as the retinoic acid–inducible early gene 1 (Rae-1) protein family and H-60 minor histocompatibility antigen in mice46. Ligation of NKG2D results in signal transduction via DAP10, leading to NK cell cytotoxicity46. HCMV encodes UL16, which in a soluble form is capable of binding ULBP, thus blocking the interaction between the NKG2D activating receptor complex and its cognate activating ligand47,48. These data suggest a mechanism by which UL16 might inhibit NK cell function in vivo.
A final means by which viruses interfere with activating receptor function is through the inhibition of signaling induced by activating-receptor ligation. Although these mechanisms can potentially inhibit many cells, at least one viral protein appears to have some specificity for NK cell activation. HIV-1 Tat can block the NK cell activation and function induced by ligation of LFA-1 on the NK cell surface49,50. Tat does not affect NK cell adhesion to the target cell, but specifically binds to an L-type calcium channel on NK cells49. Its binding blocks calcium influx and subsequent induction of the calcium-calmodulin kinase II in NK cells, which is required for cytotoxicity50. The search for viral products that specifically block signaling for NK cell cytotoxicity will certainly become more productive as the appreciation of these pathways in NK cells grows.
Evasion by modulation of cytokines or chemokines
Viruses may subvert NK cell responses through virus-encoded proteins that counteract or modulate the interactions between cytokine or chemokine molecules and their cognate receptors. Numerous poxviruses and herpesviruses encode homologs to known cytokines and chemokines with agonistic or antagonistic function, or secreted proteins or receptors that bind with high affinity to cytokines and chemokines51. Although several examples of this sort of molecular mimicry have been described for different viruses, direct evidence for the involvement of NK cells in this strategy of immune escape in vivo is scarce. Interference with anti-viral NK cell function could involve inhibition or antagonism of cytokines such as IL-12, IL-18, TNF-α, IL-1α, IL-1β and IL-15, which participate in inducing NK cell IFN-γ production and cytotoxicity8. Alternatively, viruses could facilitate overproduction or encode homologs of other cytokines (such as IL-1052) that have an inhibitory effect upon NK cells. Among the chemokines, targets for viral modulation could involve those that directly affect NK cell chemotaxis, including MIP-1α (CCL3), MIP-1β (CCL4), MCP-1 (CCL2), MCP-2 (CCL8), MCP-3 (CCL7) and RANTES (CCL5)8, or other chemokines and chemokine receptors involved in recruitment of leukocyte subsets that influence NK cell function.
Several virus-produced chemokine homologs have been described that might interfere with NK cell–mediated defense. The putative CC-chemokine homolog m131-m129 (or MCK-2) of MCMV is directly linked to NK cell evasion53,54. Infection of mice with an m131-m129–deleted mutant virus results in decreased viral burden in the spleen and liver. The increased clearance of this mutant virus is negated by NK cell depletion, suggesting that m131-m129 inhibits NK cell–mediated viral clearance. The exact mechanism underlying this function of m131-m129 is unclear. Another chemokine homolog with possible relevance to NK cells is the broad-spectrum CC, CXC and CX3C chemokine antagonist vMIP-II of KHSV, which could block chemotactic responses of monocytes to RANTES, MIP-1α and MIP-1β55. In addition, the CCR8 antagonist vMIP-I binds to chemokine receptors on NK cells56. Similarly, MCV also encodes a homolog, MC148, that is a narrow-spectrum antagonist of MCP-157. Each of these chemokine antagonists could potentially interfere with NK cell responses. In contrast, virus-encoded homologs of the cytokine IL-10, which function as agonists, have been cloned from HCMV and Epstein-Barr virus (EBV)58,59. These viral proteins may impair NK cell activation by inhibiting production of type 1 cytokines as well as by directly acting upon NK cells.
Specific interference with NK cell activation can also be mediated by cytokine-binding protein homologs. A principal target is IL-18, which is central to NK cell production of IFN-γ. Ectromelia poxvirus (EV) encodes a cytokine-binding protein with NK cell–specific effects60. The EV p13 protein is homologous to mammalian IL-18BP, binds murine IL-18 and inhibits IL-18 receptor binding and activity. Mice infected with a p13-deleted mutant virus have markedly greater NK cell cytotoxicity than those infected with wild-type EV, as a result of increased NK cell activation. Compared to mice infected with p13-deleted virus, mice infected with wild-type EV have lower IFN-γ production and higher IL-10 production, further highlighting the importance of this viral protein in NK cell evasion60. MCV also encodes several gene products that likely modulate host immunity61,62, including a functional IL-18BP homolog, MC54L63. The evolution of these homologs points to the importance of the NK cell IFN-γ pathway in the elimination of virus-infected cells, and shows the importance of IL-18 function.
Binding and sequestration of IL-18 from its cognate receptor has also been suggested to be involved in human papillomavirus (HPV)–related pathogenesis64,65. The oncoproteins HPV16 E6 and E7 inhibit IL-18–induced IFN-γ production in PHA-stimulated PBMC and in an IL-12–stimulated immortalized NK cell line by specific and competitive binding to IL-1865 or IL-18Rα64.
Other proteins without homology to known cytokine or chemokine receptors or binding proteins that can act as cytokine or chemokine antagonists include murine γ-herpesvirus 68 M3, which encodes a secreted 44-kD broad-spectrum chemokine binding protein (hvCKBP), and a 35-kD soluble CC-chemokine–specific binding protein (vCKBP) encoded by vaccinia virus. Both proteins block the activity of several chemokines, including the NK cell chemoattractants MIP-1α66,67, MCP-1 and RANTES67, and thus function as inhibitors with potential NK cell specificity.
Direct viral effects on NK cells
Viruses can exert direct effects on NK cells. In particular, viruses can infect and inhibit or destroy NK cells, or cause inhibition through presumably direct contact with NK cells. These mechanisms should be particularly advantageous to viruses that attain chronically high systemic or organ-specific titers.
Both HIV and HSV infect NK cells in vitro. HIV can infect cultured NK cells, and is found ex vivo in NK cells from HIV-seropositive individuals68,69. Although in vitro infection of NK cells with HIV does not affect the overall lytic activity of NK cell cultures, it does greatly reduce viability68. Thus, HIV may evade NK cell responses to some degree by direct infection and induction of cytopathic effects. In contrast, HSV can spread from infected fibroblasts to cultured NK cells, and thus inhibits NK cell cytotoxicity69. This direct infection of NK cells is unusual and suggests that HSV-mediated inhibition of these cells may fall into the category of direct viral effects, although the mechanism is unknown. It will be useful to ascertain whether HSV gene products can specifically interfere with intracellular NK cell signals leading to activation in infected NK cells.
Interactions between virus particles and NK cells that do not result in infection may exert other direct effects upon NK cells. One viral envelope protein has recently been implicated in inhibition of NK cells. E2, the major envelope protein of hepatitis C virus (HCV), binds to CD81 (TAPA-1), which induces a costimulatory signal in T cells70. In NK cells, however, ligation of CD81 by immobilized E2 or anti-CD81 inhibits NK cell cytotoxicity, IL-2–induced proliferation and IL-2–, IL-12– or IL-15–induced IFN-γ production71,72. In addition, CD81 ligation specifically inhibits ERK and MAPK phosphorylation induced by CD16-mediated activation of NK cells71. These effects of E2 binding to CD81 are specific to NK cells, as opposite activities are induced in T cells. Thus, E2 can mediate specific inhibition of NK cells via an inhibitory receptor, and therefore direct viral binding to NK cells may quell NK cell responses. This mechanism may be particularly useful to HCV during viremia and in the liver, where viral titers are high and NK cell defense is important.
Conclusion
Viruses have evolved numerous mechanisms to evade immune responses. Although many are used to avoid multiple immunologic effector cells, several are specifically directed at NK cells. Viral evasion strategies targeting NK cells can be divided into five mechanistic categories: ligation of inhibitory class I receptors on NK cells by virus-encoded MHC class I homologs; virus-induced regulation of class I protein expression resulting in selective expression or up-regulation of class I molecules that can bind NK cell class I inhibitory receptors; interference with NK cell–activating receptor function caused by virus-mediated inhibition of the expression of the corresponding ligand, of their signaling in infected host cells or of the production of virus encoded antagonists of NK cell–activating receptors; inhibition of NK cell activation, trafficking, or both, resulting from viral modulation of cytokine–chemokine networks or virus-encoded cytokine or chemokine homologs that prevent NK cell function; and direct inhibitory effects of viruses on NK cells, including infection of NK cells and ligation of non-class I NK cell inhibitory receptors by viral envelope proteins. These multiple mechanisms highlight the importance of NK cells in defense against viral infections. In particular, certain viruses, including herpesviruses, appear to be especially adept in their avoidance of NK cells, consistent with the crucial role of NK cells in the control of these viruses. Further study of viral evasion mechanisms will provide useful models to elucidate NK cell biology as well as to provide therapeutic targets to enhance host advantage during infection.
References
Biassoni, R. et al. Human natural killer cell receptors and co-receptors. Immunol. Rev. 181, 203–214 (2001).
Smith, H.R., Idris, A.H. & Yokoyama, W.M. Murine natural killer cell activation receptors. Immunol. Rev. 181, 115–125 (2001).
Mandelboim, O. et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 409, 1055–1060 (2001).
Arase, H., Mocarski, E.S., Campbell, A.E., Hill, A.B. & Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326 (2002).
Smith, H.R. et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 99, 8826–8831 (2002).
Melero, I. et al. Signaling through the LFA-1 leucocyte integrin actively regulates intercellular adhesion and tumor necrosis factor-α production in natural killer cells. Eur. J. Immunol. 23, 1859–1865 (1993).
Boles, K.S., Stepp, S.E., Bennett, M., Kumar, V. & Mathew, P.A. 2B4 (CD244) and CS1: novel members of the CD2 subset of the immunoglobulin superfamily molecules expressed on natural killer cells and other leukocytes. Immunol. Rev. 181, 234–249 (2001).
Biron, C.A., Nguyen, K.B., Pien, G.C., Cousens, L.P. & Salazar-Mather, T.P. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17, 189–220 (1999).
Biron, C.A., Byron, K.S. & Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 320, 1731–1735 (1989).
Ballas, Z.K., Turner, J.M., Turner, D.A., Goetzman, E.A. & Kemp, J.D. A patient with simultaneous absence of “classical” natural killer cells (CD3−, CD16+, and NKH1+) and expansion of CD3+, CD4−, CD8−, NKH1+ subset. J. Allergy Clin. Immunol. 85, 453–459 (1990).
Wendland, T., Herren, S., Yawalkar, N., Cerny, A. & Pichler, W.J. Strong αβ and γδ TCR response in a patient with disseminated mycobacterium avium infection and lack of NK cells and monocytopenia. Immunol. Lett. 72, 75–82 (2000).
Beck, S. & Barrell, B.G. Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature 331, 269–272 (1988).
Reyburn, H.T. et al. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature 386, 514–517 (1997).
Braud, V.M. et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 391, 795–799 (1998).
Tomasec, P. et al. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 287, 1031 (2000).
Leong, C.C. et al. Modulation of natural killer cell cytotoxicity in human cytomegalovirus infection: the role of endogenous class I major histocompatibility complex and a viral class I homolog. J. Exp. Med. 187, 1681–1687 (1998).
Cosman, D. et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 7, 273–282 (1997).
Farrell, H.E. et al. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature 386, 510–514 (1997).
Kubota, A., Kubota, S., Farrell, H.E., Davis-Poynter, N. & Takei, F. Inhibition of NK cells by murine CMV-encoded class I MHC homologue m144. Cell. Immunol. 191, 145–151 (1999).
Cretney, E. et al. m144, a murine cytomegalovirus (MCMV)-encoded major histocompatibility complex class I homologue, confers tumor resistance to natural killer cell-mediated rejection. J. Exp. Med. 190, 435–444 (1999).
Kloover, J.S., Grauls, G.E., Blok, M.J., Vink, C. & Bruggeman, C.A. A rat cytomegalovirus strain with a disruption of the r144 MHC class I-like gene is attenuated in the acute phase of infection in neonatal rats. Arch. Virol. 147, 813–824 (2002).
Senkevich, T.G. & Moss, B. Domain structure, intracellular trafficking, and β2-microglobulin binding of a major histocompatibility complex class I homolog encoded by molluscum contagiosum virus. Virology 250, 397–407 (1998).
Tortorella, D., Gewurz, B.E., Furman, M.H., Schust, D.J. & Ploegh, H.L. Viral subversion of the immune system. Annu. Rev. Immunol. 18, 861–926 (2000).
Gewurz, B.E., Wang, E.W., Tortorella, D., Schust, D.J. & Ploegh, H.L. Human cytomegalovirus US2 endoplasmic reticulum-lumenal domain dictates association with major histocompatibility complex class I in a locus-specific manner. J. Virol. 75, 5197–5204 (2001).
Machold, R.P., Wiertz, E.J., Jones, T.R. & Ploegh, H.L. The HCMV gene products US11 and US2 differ in their ability to attack allelic forms of murine major histocompatibility complex (MHC) class I heavy chains. J. Exp. Med. 185, 363–366 (1997).
Schust, D.J., Tortorella, D., Seebach, J., Phan, C. & Ploegh, H.L. Trophoblast class I major histocompatibility complex (MHC) products are resistant to rapid degradation imposed by the human cytomegalovirus (HCMV) gene products US2 and US11. J. Exp. Med. 188, 497–503 (1998).
Lopez-Botet, M., Llano, M. & Ortega, M. Human cytomegalovirus and natural killer-mediated surveillance of HLA class I expression: a paradigm of host-pathogen adaptation. Immunol. Rev. 181, 193–202 (2001).
Jun, Y. et al. Human cytomegalovirus gene products US3 and US6 down-regulate trophoblast class I MHC molecules. J. Immunol. 164, 805–811 (2000).
Park, B. et al. The MHC class I homolog of human cytomegalovirus is resistant to down-regulation mediated by the unique short region protein (US)2, US3, US6, and US11 gene products. J. Immunol. 168, 3464–3469 (2002).
Kavanagh, D.G., Gold, M.C., Wagner, M., Koszinowski, U.H. & Hill, A.B. The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J. Exp. Med. 194, 967–978 (2001).
Cohen, G.B. et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10, 661–671 (1999).
Le Gall, S. et al. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 8, 483–495 (1998).
Swigut, T., Iafrate, A.J., Muench, J., Kirchhoff, F. & Skowronski, J. Simian and human immunodeficiency virus Nef proteins use different surfaces to downregulate class I major histocompatibility complex antigen expression. J. Virol. 74, 5691–5701 (2000).
Coscoy, L., Sanchez, D.J. & Ganem, D. A novel class of herpesvirus-encoded membrane-bound E3 ubiquitin ligases regulates endocytosis of proteins involved in immune recognition. J. Cell Biol. 155, 1265–1273 (2001).
Ishido, S., Wang, C., Lee, B.S., Cohen, G.B. & Jung, J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74, 5300–5309 (2000).
Ishido, S. et al. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi's sarcoma-associated herpesvirus K5 protein. Immunity 13, 365–374 (2000).
Ulbrecht, M. et al. Cutting edge: the human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J. Immunol. 164, 5019–5022 (2000).
Braud, V., Jones, E.Y. & McMichael, A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur. J. Immunol. 27, 1164–1169 (1997).
Wang, E.C. et al. UL40-mediated NK evasion during productive infection with human cytomegalovirus. Proc. Natl. Acad. Sci. USA 99, 7570–7575 (2002).
Pietra, G. et al. Natural killer cells lyse autologous herpes simplex virus infected targets using cytolytic mechanisms distributed clonotypically. J. Med. Virol. 62, 354–363 (2000).
Huard, B. & Fruh, K. A role for MHC class I down-regulation in NK cell lysis of herpes virus-infected cells. Eur. J. Immunol. 30, 509–515 (2000).
Fletcher, J.M., Prentice, H.G. & Grundy, J.E. Natural killer cell lysis of cytomegalovirus (CMV)-infected cells correlates with virally induced changes in cell surface lymphocyte function-associated antigen-3 (LFA-3) expression and not with the CMV-induced down-regulation of cell surface class I HLA. J. Immunol. 161, 2365–2374 (1998).
Krmpotic, A. et al. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 3, 529–535 (2002).
Coscoy, L. & Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest. 107, 1599–1606 (2001).
Zheng, Z.Y. & Zucker-Franklin, D. Apparent ineffectiveness of natural killer cells vis-a-vis retrovirus-infected targets. J. Immunol. 148, 3679–3685 (1992).
Sutherland, C.L., Chalupny, N.J. & Cosman, D. The UL16-binding proteins, a novel family of MHC class I-related ligands for NKG2D, activate natural killer cell functions. Immunol. Rev. 181, 185–192 (2001).
Kubin, M. et al. ULBP1, 2, 3: novel MHC class I-related molecules that bind to human cytomegalovirus glycoprotein UL16, activate NK cells. Eur. J. Immunol. 31, 1428–1437 (2001).
Cosman, D. et al. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14, 123–133 (2001).
Zocchi, M.R., Rubartelli, A., Morgavi, P. & Poggi, A. HIV-1 Tat inhibits human natural killer cell function by blocking L-type calcium channels. J. Immunol. 161, 2938–2943 (1998).
Poggi, A. et al. NK cell activation by dendritic cells is dependent on LFA-1-mediated induction of calcium-calmodulin kinase II: inhibition by HIV-1 Tat C-terminal domain. J. Immunol. 168, 95–101 (2002).
Lalani, A.S., Barrett, J.W. & McFadden, G. Modulating chemokines: more lessons from viruses. Immunol. Today 21, 100–106 (2000).
D'Andrea, A. et al. Interleukin 10 (IL-10) inhibits human lymphocyte interferon γ production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med. 178, 1041–1048 (1993).
Fleming, P. et al. The murine cytomegalovirus chemokine homolog, m131/129, is a determinant of viral pathogenicity. J. Virol. 73, 6800–6809 (1999).
Saederup, N., Aguirre, S.A., Sparer, T.E., Bouley, D.M. & Mocarski, E.S. Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J. Virol. 75, 9966–9976 (2001).
Kledal, T.N. et al. A broad-spectrum chemokine antagonist encoded by Kaposi's sarcoma–associated herpesvirus. Science 277, 1656–1659 (1997).
Inngjerdingen, M., Damaj, B. & Maghazachi, A.A. Expression and regulation of chemokine receptors in human natural killer cells. Blood 97, 367–375 (2001).
Luttichau, H.R. et al. A highly selective CC chemokine receptor (CCR) 8 antagonist encoded by the poxvirus molluscum contagiosum. J. Exp. Med. 191, 171–180 (2000).
Kotenko, S.V., Saccani, S., Izotova, L.S., Mirochnitchenko, O.V. & Pestka, S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc. Natl Acad. Sci. USA 97, 1695–1700 (2000).
Moore, K.W. et al. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science 248, 1230–1234 (1990).
Born, T.L. et al. A poxvirus protein that binds to and inactivates IL-18, and inhibits NK cell response. J. Immunol. 164, 3246–3254 (2000).
Senkevich, T.G. et al. Genome sequence of a human tumorigenic poxvirus: prediction of specific host response–evasion genes. Science 273, 813–816 (1996).
Senkevich, T.G., Koonin, E.V., Bugert, J.J., Darai, G. & Moss, B. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 233, 19–42 (1997).
Xiang, Y. & Moss, B. IL-18 binding and inhibition of interferon γ induction by human poxvirus-encoded proteins. Proc. Natl Acad. Sci. USA 96, 11537–11542 (1999).
Lee, S.J. et al. Both E6 and E7 oncoproteins of human papillomavirus 16 inhibit IL-18-induced IFN-γ production in human peripheral blood mononuclear and NK cells. J. Immunol. 167, 497–504 (2001).
Cho, Y.S. et al. Down modulation of IL-18 expression by human papillomavirus type 16 E6 oncogene via binding to IL-18. FEBS Lett. 501, 139–145 (2001).
Parry, C.M. et al. A broad spectrum secreted chemokine binding protein encoded by a herpesvirus. J. Exp. Med. 191, 573–578 (2000).
Alcami, A., Symons, J.A., Collins, P.D., Williams, T.J. & Smith, G.L. Blockade of chemokine activity by a soluble chemokine binding protein from vaccinia virus. J. Immunol. 160, 624–633 (1998).
Chehimi, J. et al. In vitro infection of natural killer cells with different human immunodeficiency virus type 1 isolates. J. Virol. 65, 1812–1822 (1991).
York, I.A. & Johnson, D.C. Direct contact with herpes simplex virus-infected cells results in inhibition of lymphokine-activated killer cells because of cell-to-cell spread of virus. J. Infect. Dis. 168, 1127–1132 (1993).
Wack, A. et al. Binding of the hepatitis C virus envelope protein E2 to CD81 provides a co-stimulatory signal for human T cells. Eur. J. Immunol. 31, 166–175 (2001).
Crotta, S. et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med. 195, 35–41 (2002).
Tseng, C.T. & Klimpel, G.R. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 195, 4349 (2002).
Acknowledgements
We thank R. Hellmiss for graphic support and H. L. Ploegh for his review of this manuscript. Supported by US National Institutes of Health grants AI-50207 (to J. L. S.) and AI-07512 (to J. S. O.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Orange, J., Fassett, M., Koopman, L. et al. Viral evasion of natural killer cells. Nat Immunol 3, 1006–1012 (2002). https://doi.org/10.1038/ni1102-1006
Issue Date:
DOI: https://doi.org/10.1038/ni1102-1006
This article is cited by
-
Pestaloamides A and B, two spiro-heterocyclic alkaloid epimers from the plant endophytic fungus Pestalotiopsis sp. HS30
Science China Chemistry (2020)
-
How I Manage Natural Killer Cell Deficiency
Journal of Clinical Immunology (2020)
-
Avian influenza virus directly infects human natural killer cells and inhibits cell activity
Virologica Sinica (2017)
-
Peginterferon Alfa-2a/Ribavirin treatment efficacy in chronic hepatitis C patients is related to natural killer group 2D gene rs1049174 GC polymorphism
VirusDisease (2016)
-
Natural killer cell response to BK virus infection in polyoma virus–associated nephropathy of renal transplant recipients
Kidney International (2013)
