
Abstract
The importance of immunoproteasomes to antigen presentation has been unclear because animals totally lacking immunoproteasomes had not been available. Having now developed mice lacking the three immunoproteasome catalytic subunits, we found that the dendritic cells of these mice had defects in presenting several major histocompatibility complex (MHC) class I epitopes. During viral infection in vivo, the presentation of a majority of MHC class I epitopes was markedly reduced in immunoproteasome-deficient animals compared with wild-type animals, whereas presentation of MHC class II peptides was unaffected. According to mass spectrometry, the repertoire of MHC class I–presented peptides was ∼50% different from that in wild-type mice, and these differences were sufficient to stimulate robust transplant rejection of wild-type cells in mutant mice. These results indicated that immunoproteasomes were more important in antigen presentation than previously thought.
Similar content being viewed by others


Main
Proteasomes are vital in generating peptides for presentation on MHC class I molecules1. Each proteasome consists of 14 structural subunits and 6 catalytic subunits (two each of the β1, β2 and β5 subunits)2. In addition to these three catalytic subunits, three alternative catalytic subunits denoted β1i (LMP2 or Psmb9), β2i (MECL1 or Psmb10) and β5i (LMP7 or Psmb8) are constitutively expressed in a number of hematopoietic cells and are induced in other cell types by interferon-γ (IFN-γ)2,3. When expressed, these alternative subunits are preferentially incorporated into newly assembling complexes to form immunoproteasomes3 and change the catalytic activities of these complexes. Compared with the constitutive proteasomes, immunoproteasomes cleave more rapidly after hydrophobic and basic amino acid residues and less rapidly after acidic ones4,5,6. As peptides with hydrophobic or sometimes basic C termini preferentially bind to MHC class I molecules7, it has long been suggested that immunoproteasomes have a specialized role in creating antigenic peptides. However, mice lacking individual immunoproteasome catalytic subunits have relatively modest changes in antigen presentation. For example, β5i-deficient mice have moderately (∼50%) lower MHC class I surface expression8 and lower or higher efficiency in presenting only a few epitopes, whereas the majority of immunogenic peptides examined are presented normally8,9,10,11,12,13,14,15. The published analyses have examined only the presentation of known epitopes, and it is unknown whether and how often immunoproteasome-deficient mice present different peptides compared with wild-type mice.
To determine whether the modest changes in these mice were due to some contribution from the remaining immunoproteasome catalytic subunits, we created a mouse that was triply deficient, lacking all three immune subunits. Because the genes encoding β1i (Psmb9) and β5i (Psmb8) are so close together on chromosome 17, which made the chance of generating a doubly deficient mouse by crossing β5i-null with β1i-null mice vanishingly small, we chose to create a new sequential deletion of these two genes. We then bred the β1i and β5i doubly deficient mice to β2i-deficient (Psmb10-null) mice to create the animal with a triple immunoproteasome deficiency. We found that the triply deficient mice had altered presentation of most of the epitopes we tested, both in vitro and in vivo, and that these changes in antigen presentation were sufficient to cause triply deficient mice to reject wild-type cells.
Results
Generation of immunoproteasome–triply deficient mice
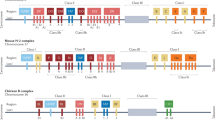
To generate the β1i β5i doubly deficient animals, we designed a sequential deletion strategy (Fig. 1a). First a LacZ-FRT-neo-FRT construct was fused in frame to the start codon (27 base pairs (bp) downstream of the 5′ end of exon 1) of Psmb8 (which encodes β5i), removing the remainder of exon 1 plus exons 2 through 5 by homologous recombination. The neo gene was then removed by enhanced FLP recombinase (FLPe) activity in the bacteria. An alkaline phosphatase–loxP-neo-loxP construct was then fused in frame to the start codon (22 bp from the 5′ end of exon 1) of Psmb9 (which encodes β1i), removing the remainder of exon 1 by homologous recombination. The doubly deficient mouse was created on a 129 background, and mice were back-crossed nine times onto the C57BL/6J background. These mice were then bred to β2i-null16 mice to yield mice triply deficient in the immunoproteasome. By the end of this process, the resulting animals had been backcrossed ten generations onto the C57BL/6 background. The progeny of these animals were analyzed for 110 single-nucleotide polymorphisms spanning the genome and were found to be fully backcrossed to C57BL/6.

(a) Exons targeted for the sequential deletion of both Psmb9 and Psmb8 to generate doubly deficient mice. (b,c) Immunoblots showing that β1i and β5i proteins were not detected in spleen cell lysates (b) or proteasomes purified from splenocytes (c) of triple-knockout (TKO) mice (equivalent protein loaded in each lane). WT, wild-type. (d) Immunoblot showing that the amount of proteasomes in WT and TKO spleens was similar, as measured by α subunits in serial dilutions of proteasome pellets from equal numbers of spleens. (e) Normalized Tap1 mRNA expression in WT and β5i β1i double-knockout (DKO) splenocytes, determined by RT-qPCR. *P < 0.05 (two-tailed, unpaired t-test). Error bars indicate mean ± s.d. from five animals for each strain. (f) Immunoblotting of splenocyte whole-cell lysates from indicated strains with antibody to Tap1 (equivalent protein loaded in each lane). Data are representative of two (b–e) or four (f) experiments.
The β1i and β5i subunits were undetectable by immunoblotting in lysates or proteasome preparations from the immunoproteasome-deficient spleens, as expected (Fig. 1b,c). Also as expected, the total amount of proteasomes was the same in the triply deficient and wild-type spleens, as assessed by immunoblotting for an invariant α subunit in proteasome preparations (Fig. 1d). We also found that the amount of ubiquitinated proteins did not differ between the triply deficient and wild-type cells (Supplementary Fig. 1).
Because the Psmb8 and Psmb9 genes flank the transporter 1, ATP-binding cassette, sub-family B (MDR/TAP) gene (Tap1), we evaluated the expression of Tap1 in the β1i β5i doubly deficient mice to see if it was affected. The expression of Tap1 mRNA (Fig. 1e) and protein (Fig. 1f) were ∼50% lower than in wild-type mice. The amount of Tap1 protein expressed in the doubly deficient mice was similar to that of Tap1+/− heterozygous mice (Fig. 1f), and therefore we used the latter mice as controls in some experiments.
The triply deficient mice were viable and fertile, and appeared healthy. The overall cellularities of their thymuses and spleens were not different from those of wild-type animals (Supplementary Fig. 2a,b). However, there were ∼50% fewer mature CD8+ T cells in the immunoproteasome-deficient thymus and spleen (Supplementary Fig. 2c,d), which was similar to what has been observed in β1i singly deficient mice9. There was no difference in the number of B cells in wild-type and triply deficient mice, which, although different from observations in β1i-deficient mice11,17, was similar to findings in β1i β2i doubly deficient mice17.
MHC class I expression in triply deficient mice
MHC class I molecules must bind peptides to be released from the endoplasmic reticulum and trafficked to the cell surface18. Consequently, the surface expression of MHC class I molecules is a proxy for the overall supply of peptides for antigen presentation. Therefore, we evaluated the surface expression of MHC class I molecules in the mutant mice by using flow cytometry to analyze cells stained with antibodies to MHC class I molecules. Cells lacking all three immunoproteasome catalytic subunits had only ∼50% of the MHC class I surface expression of wild-type cells (Fig. 2a,b). Both H-2Kb and H-2Db molecules were similarly affected. There was less MHC class I surface expression on CD4+ T cells and CD8+ T cells in the blood, spleen, lymph nodes and thymus of triply deficient mice, as well as on lymph node B cells (B220+ cells) and splenic macrophages (CD11b+ splenocytes), dendritic cells (DCs; CD11c+ splenocytes) and B cells (B220+ splenocytes). These findings were similar to the expression in cells lacking only β5i, whereas cells lacking β1i or β2i expressed MHC class I molecules more similarly to wild-type cells (Fig. 2c,d), as reported8,9,16. We found no defect in MHC class I surface expression in Tap1+/− T or B cells, although we did see slightly less H2-Kb expression in splenic DCs. Therefore, the lower MHC surface expression on the triply deficient cells was not due to their modestly lower Tap1 expression.

WT, wild-type; TKO, triple knockout. (a,b) H-2Kb (a) and H-2Db (b) surface expression on cells from WT and TKO mice. LN, lymph nodes. In both cases, MHC class I surface expression in TKO mice was significantly lower than in WT mice (P < 0.0001, two-way ANOVA, n between 3 and 33 depending on the strain and tissue). Error bars indicate mean ± s.d. (c,d) H-2Kb surface expression on splenic B cells (splenocytes gated on B220+; c) or DCs (splenocytes gated on CD11c+; d). Scatterplots represent individual animals, with mean ± s.d. indicated. *P < 0.05 (one-way ANOVA followed by Dunnett's multiple-comparison test, n = 3 to 6 for each strain). All results are normalized to the average from wild-type controls in each experiment. Data are combined from ten (a,b) or two (c,d) experiments.
To assay the relative stability of peptide–MHC class I (pMHCI) complexes at the surface of triply deficient cells, we treated wild-type and triply deficient splenocytes with brefeldin A (BFA) for 4 h and monitored the abundance of MHC class I molecules on the cell surface. We found no evidence that pMHCI complexes (H-2Kb or H-2Db) were less stable in triply deficient animals (Supplementary Fig. 3). The lower surface expression, therefore, was most probably the result of less export of mature pMHCI complexes as opposed to more degradation of MHC class I molecules unstably bound to suboptimal peptides.
Defects in in vitro antigen presentation
Because the surface expression of MHC class I was lower on a broad range of lymphoid and myeloid cells, we inferred that there were defects in antigen processing that limited the overall supply of peptides to MHC class I molecules. To evaluate antigen presentation more directly, we examined the presentation of a number of well-characterized epitopes by these cells. We used DCs for many of these assays because they have been shown to constitutively express the immunoproteasome catalytic subunits19,20.
To examine presentation of Smcy738–746, a peptide antigen derived from the male H-Y antigen, we measured proliferation of Smcy-specific (purified from H-Y T cell antigen receptor (TCR) transgenic mice)21,22 in response to male C57BL/6 and triply deficient bone marrow–derived DCs (BMDCs). The proliferation of H-Y T cells cultured with male (antigen-bearing) triply deficient DCs was much less than those cultured with wild-type DCs, and was similar to that of H-Y T cells cultured with DCs from C57BL/6 females, which lack the Smcy antigen (Fig. 3a). In contrast, the presentation of the Smcy epitope by DCs from mice singly deficient in any of the three immunoproteasome subunits or from Tap1+/− mice was similar to that seen with wild-type DCs.

(a) Proliferation (measured by [3H]thymidine incorporation; c.p.m., counts per million) of H-Y T cells in response to antigen presentation by BDMCs from indicated mouse strains (male unless otherwise noted). WT, wild-type; TKO, triple knockout. (b) Uty antigen presentation in male BMDCs of the indicated strains, assayed with 11p9z hybridoma cells. Secreted IL-2 was measured with the CTLL bioassay. (c) NP366 antigen presentation by influenza-infected wild-type and triply deficient BMDCs assayed with 12.64-CD8αβ-LUC hybridoma. RLU, relative luciferase units. (d) SIINFEKL antigen presentation by irradiated splenocytes from WT, OVA-transgenic (Tg) or TKO OVA-transgenic animals, assayed with RF33.70-LUC hybridoma cells. (e) SIINFEKL presentation in WT and TKO BMDCs infected with rVV-SIINFEKL and assayed with RF33.70-LUC hybridoma cells. In a–d, responses to TKO cells were significantly different from responses to WT cells (P < 0.0001, two-way ANOVA); responses were not significantly different in e. Error bars indicate s.d. of triplicate values. Data are representative of four (a,c), three (b) or two (d,e) experiments.
In addition, we measured the presentation of the male antigen Uty246–254 to the T-T hybridoma 11p9z (refs. 23,24). Triply deficient DCs also had severely impaired presentation of this epitope (Fig. 3b). Consistent with published results13, we found that β5i singly deficient DCs also showed defective presentation in this assay.
To further assess potential deficits in antigen processing and presentation by triply deficient cells, we used two other systems. First, we measured presentation of influenza peptide NP366 by influenza-infected BMDCs to the 12.64-CD8αβ-LUC hybridoma (a derivative of 12.64 (ref. 25) that expresses firefly luciferase under the control of the NFAT promoter) and found that triply deficient BMDCs presented this epitope much more poorly than wild-type cells, especially at early time points (Fig. 3c). Published reports have found no effect of β2i deficiency or β5i β2i double deficiency14 on NP366 presentation, but the reports have differed as to whether β1i single deficiency affects NP366 presentation (findings have varied from no effect to a partial reduction)9,10,14.
Second, we measured presentation of the OVA257–264 epitope (SIINFEKL) to the RF33.70-LUC hybridoma (a derivative of RF33.70 (ref. 26) that expresses firefly luciferase under the control of the NFAT promoter) by splenocytes from OVA transgenic mice with intact immunoproteasomes and from triply deficient OVA transgenic mice. We found that the triply deficient spleens presented less of this epitope than wild-type cells (Fig. 3d). In contrast, we found that when cells were infected with recombinant vaccinia virus expressing a minigene that did not require cleavage (rVV-SIINFEKL), presentation of SIINFEKL was not reduced in triply deficient cells (Fig. 3e). Published studies have shown that β1i-deficient9, β5i-deficient12 and β5i β2i doubly deficient animals15 present this antigen normally. Overall, these analyses revealed that cells lacking immunoproteasomes had a broad defect in antigen presentation that affected all four epitopes examined.
Reduced presentation of LCMV epitopes in vivo
To measure antigen presentation in vivo, we transferred wild-type P14 TCR-transgenic T cells (specific for GP33 viral epitopes) into wild-type or triply deficient mice infected with lymphochoriomeningitis virus (LCMV) and assayed the stimulation of the T cells. Two days after infection with LCMV, wild-type and triply deficient mice were injected with naïve BFA-treated P14 lymphocytes (and, in addition, BFA to maintain the secretion block). Splenocytes were then harvested 4 h later from these animals, and the production of tumor necrosis factor (TNF) and IFN-γ by the P14 cells was detected by intracellular cytokine staining and quantified by flow cytometry. This protocol allowed us to quantify activation of the TCR-transgenic T cells by their cognate antigen within the first 4 h of stimulation by antigen-presenting cells in vivo. Of note, there was no relative loss of the P14 T cells transferred into triply deficient mice because the time point we analyzed was much too early for immune-based rejection to occur (an issue discussed below). Production of both TNF and IFN-γ by P14 cells was significantly lower in triply deficient mice than in wild-type controls (P < 0.05; Fig. 4a,b), consistent with less presentation of the GP33 epitope in triply deficient hosts.

(a,b) Flow cytometry results showing stimulation of T cells from spleens of wild-type and triply deficient mice injected 2 d after LMCV infection with BFA and BFA-treated P14-transgenic splenocytes; graphs show percentage of P14 T cells producing TNF and IFN-γ. Scatterplots represent individual animals, with mean ± s.d. indicated. (c,d) Presentation of LCMV antigens in vivo. Spleens were harvested 5.5 d after infection and cells were analyzed by in vitro re-stimulation followed by surface and intracellular cytokine staining. Error bars show mean and s.d. from three animals of each strain. Inset in c, low-abundance CD8+ T cell responses. *P < 0.05 (two-tailed, unpaired t-test). Data are representative of three (a,b) or two (c,d) experiments. Med, medium alone, without peptide.
To evaluate the role of immunoproteasomes in the presentation of other LCMV antigens in vivo, we transferred LCMV-immune wild-type splenocytes into wild-type and triply deficient mice, and 24 h later infected the animals with LCMV. To prevent rejection of the adoptively transferred cells, we depleted the host T cells with antibody to Thy1.2, and, as expected, there was no relative loss of the transferred cells in triply deficient hosts (see below). We then harvested splenocytes from wild-type and triply deficient hosts 5.5 d after infection and quantified the activation of different peptide-specific T cells by in vitro re-stimulation and intracellular cytokine staining. CD8+ T cell responses to GP33 were lower in triply deficient hosts than in wild-type hosts, confirming the results obtained with the P14-transgenic T cells, which recognize this peptide. Moreover, responses to most other epitopes in vivo were significantly lower (P < 0.05; Fig. 4c), consistent with a defect in processing and presentation of a broad array of epitopes in the triply deficient mice. Although it is formally possible that the responses of wild-type T cells in triply deficient animals could have been affected by host factors, such as regulatory T cells, the weaker responses of the transferred T cells in triply deficient animals most probably reflected reduced antigen presentation, especially because the host was depleted of T cells. Furthermore, as the responding T cells were wild type, any changes in the repertoire of immunoproteasome-deficient T cells or any defect in their proliferative responses would not skew the results (although we did not observe any such defects in responses of triply deficient T cells to antigens (Supplementary Fig. 4) or in homeostatic proliferation during two experiments in which these cells were transferred to lymphopenic RAG-deficient hosts (data not shown)).
Responses to three peptides, NP396, GP276 and GP92, were not weaker in triply deficient hosts, and the response to NP205 was weaker but did not reach the level of statistical significance (P > 0.05; Fig. 4c). In the case of the NP396 epitope, it is possible that our assay system could have missed a reduction in its presentation if the number of peptide-MHC complexes was in excess of what was needed to maximally stimulate the T cells; this might occur in a viral infection because the NP396 peptide has a very high binding affinity for H-2Db (ref. 27). The LCMV GP276 epitope, in contrast, seemed to be produced better by the triply deficient animals. This epitope has been shown to be destroyed by β1i in vitro28, so its greater presentation may have reflected less destruction of this epitope in triply deficient antigen-presenting cells (APCs).
Given that the responses to GP33 and GP34 were weaker in triply deficient mice whereas the responses to NP396 were not significantly different, the immunodominance hierarchy was markedly altered in the triply deficient mice. The weaker T cell responses in triply deficient mice were substantially more widespread than what has been reported in singly deficient animals. β1i-deficient11 and β2i-deficient16 mice have been shown to have weaker CD8+ T cell responses to LCMV GP276 and NP205, but normal responses to NP396, GP33 and several other LCMV epitopes. β5i-deficient mice have been reported to have normal responses to all LCMV peptides11.
We did not find weaker responses of CD4+ T cells to the MHC class II epitopes GP61 and NP309 in triply deficient mice (Fig. 4d), although the NP309 responses were very close to background in both wild-type and triply deficient animals. We also examined CD4+ T cell responses in unmanipulated wild-type and triply deficient mice infected with LCMV; we found that the responses to these epitopes were not weaker in triply deficient mice (Supplementary Fig. 4) and that the NP309 response was easier to detect above the background in these experiments. The normal CD4+ T cell responses in LCMV-infected triply deficient mice suggested that neither APCs nor T cells were globally affected in these mice, but that the defects were specific to MHC class I antigen presentation.
Triply deficient mice reject wild-type splenocytes
The finding that immunoproteasome-deficient APCs had marked differences from wild-type cells in the presentation of a majority of tested T cell epitopes raised the possibility that these mutant cells might not only have quantitative changes in antigen presentation but also be generating a different set of peptides. If this were the case, triply deficient T cells would not be tolerant to many of the self peptides presented on immunoproteasome-expressing wild-type cells and would be stimulated to reject these cells. To test this hypothesis, we injected triply deficient mice with a mixture of wild-type and triply deficient splenocytes, each group labeled with different amounts of the cytosolic dye CFSE, and then monitored the proportion of wild-type to triply deficient splenocytes in the blood of host animals over 11 to 12 d. Around day 4 after transfer, the wild-type cells progressively disappeared in triply deficient hosts and were profoundly depleted by 11 d after injection (Fig. 5a). We also performed these experiments with the three singly deficient strains and found that wild-type cells were also rejected by the singly deficient animals, albeit with much slower kinetics (Fig. 5a,b). These results suggested that wild-type splenocytes presented a markedly different repertoire of peptides than those found in either the thymus or periphery of triply deficient animals, resulting in recognition of this immunoproteasome-derived repertoire as foreign epitopes. Moreover, the data were consistent with a defect in the generation of presented peptides in triply deficient mice that was substantially greater than the defect in any of the mice deficient in a single immunoproteasome subunit. To confirm that the recognition and rejection of the wild-type cells was mediated by CD8+ T cells, we depleted host T cells using a Thy1.2-specific antibody. Triply deficient animals whose T cells had been depleted did not reject wild-type splenocytes (Fig. 5c).

(a) Selective killing of wild-type (WT) cells in blood from mice of the indicated strains, were injected with WT and triple-knockout (TKO) cells differentially labeled with CFSE. Shown are mean ± s.d. from 14 mice in 3 experiments (TKO), 10 mice in 3 experiments (β1i), 5 mice in 1 experiment (β5i) and 7 mice in 2 experiments (β2i). (b) Killing of wild-type splenocytes by singly and triply deficient mice at day 7 or 8. P < 0.05 (one-way ANOVA with Dunnett's multiple-comparison test). Data are representative of QQ experiments. (c) Selective killing in a TKO host, measured as in a except that T cells were depleted by antibody to Thy1.2 (30H12) before injection of wild-type and TKO splenocytes. Control, TKO host with no anti-Thy1.2 treatment. Data are representative of two experiments.
In reciprocal experiments, we injected a mixture of CFSE-labeled wild-type and triply deficient splenocytes into wild-type mice. In contrast to the results observed in the triply deficient host, both cell populations survived in wild-type hosts (Supplementary Fig. 5). This difference between the wild-type hosts and triply deficient hosts did not surprise us. The triply deficient host had never been exposed to any of the distinct peptides generated by immunoproteasomes and therefore would not be tolerant to them. In contrast, wild-type mice expressed constitutive proteasomes and immunoproteasomes, so the animals would be expected to be tolerant to the peptides generated by both types of proteolytic complex.
Changes in MHC I peptide repertoire
Having found a difference in the peptides presented by wild-type and triply deficient splenocytes as measured by immune-based rejection, we wanted to look in more detail at the peptides bound to MHC class I in the two strains. To identify naturally processed peptides, we isolated H-2Kb or H-2Db molecules from splenocytes, eluted the peptides, fractionated them by HPLC and analyzed the results by mass spectrometry. Because triply deficient splenocytes presented approximately half as much MHC class I on the cell surface as wild-type splenocytes, we needed to use more triply deficient spleens to achieve similar yields of peptides from the two strains. In the raw (unvalidated) data from two independent experiments, we identified 398 distinct peptide sequences in the material extracted from wild-type H-2Kb samples (81% of which were not found in triply deficient cells), 581 sequences from wild-type H-2Db samples (78% not found in triply deficient cells), 331 sequences from triply deficient H-2Kb samples (77% not found in wild-type cells) and 570 sequences from triply deficient H-2Db samples (78% not found in wild-type cells). These data represented unvalidated masses, and on the basis of our previous experience we expected that they would include a large number of erroneously assigned sequences. Valid peptide sequences were identified with high confidence in an initial search with Spectrum Mill algorithm followed by expert manual spectral validation. We identified a total of 270 wild-type and 297 triply deficient validated peptide sequences. Although some validated peptides were shared by the two strains, we identified a large proportion of peptides that were found only in one strain (Fig. 6 and Supplementary Tables 1–4). Although wild-type mice can express both the constitutive and immune proteolytic subunits, wild-type splenocytes express only small amounts of the constitutive subunits29, so the peptides unique to the triply deficient splenocytes were presumably the products of constitutive proteasomes. The mass spectrometry technique that we used did not allow us to determine the relative frequency of each peptide in each strain, so our data probably underestimated the differences between the peptide repertoires of the two strains. The results showed that despite the transport selectivity of Tap1 and the binding selectivity of the MHC I molecules themselves, the differences in the biochemical activities of the proteasome and immunoproteasome subunits resulted in different peptides bound to MHC class I.

pMHCI complexes were isolated with antibodies to H-2Db (a) or H-2Kb (b). Venn diagrams show numbers of identified peptides that were unique to wild-type splenocytes, unique to triply deficient splenocytes or shared by both.
Discussion
Our results with triply deficient mice revealed the vital role of immunoproteasomes in creating peptides for MHC class I antigen presentation. The profound defects in antigen presentation that we found in the triply deficient APCs, both in vivo and in vitro, were qualitatively and quantitatively much broader than the defects in β1i, β2i or β5i singly deficient animals described by published reports and were far greater than the sum of these defects in singly deficient animals. The findings suggested that functional overlap between the subunits has masked the true importance of immunoproteasomes.
Of the 11 MHC class I presented antigens we examined, we found clear changes in 9, 8 of which had lower presentation. In the case of the Smcy antigen, expressed on male cells, there was a substantial reduction in presentation by triply deficient cells even though none of the singly deficient mice showed a defect. In the case of LCMV GP33 and GP118, there was a weakened response to these epitopes, although no such effects have been described in published studies with singly deficient animals11,16. Similarly, there was a partial defect in OVA257–264 presentation not seen in singly deficient cells9,12,15 and also a much more marked defect in the presentation of influenza NP366-374 (refs. 9,10,14). We believe, therefore, that reports on singly deficient animals have substantially underestimated the contribution of immunoproteasomes to generating peptides for MHC class I antigen presentation.
Our finding that many epitopes were presented poorly by cells that completely lack immunoproteasomes was consistent with inefficient generation of optimal peptides by constitutive proteasomes. Presumably this defect in generating peptides accounted for the approximately 50% decrease in MHC class I surface expression in the triply deficient mice. Although this decrease in MHC class I was similar to that in β5i singly deficient animals (Fig. 2 and ref. 8), the peptides that the two strains presented were almost certainly not identical. Compared with β5i-null animals, triply deficient mice presented many peptides more poorly, and they more rapidly rejected wild-type cells. The profound difference in Smcy presentation between β5i-deficient DCs (which presented the antigen normally) and triply deficient DCs (which presented the antigen very poorly), despite the quantitatively similar MHC class I surface expression, suggested that the reduced presentation in the triply deficient cells could not have been due to fewer overall MHC molecules but rather resulted from a poor supply of peptides.
In the absence of immunoproteasomes, there were changes in antigen presentation in vitro in dendritic cells and, notably, also in vivo. During LCMV infection, we observed changes that affected the magnitude of T cell responses, generally substantially weakening these responses. GP276, the one epitope for which we found better presentation by triply deficient cells, has been shown to be destroyed by β1i in vitro28. The fact that these changes in antigen presentation affected CD8+ T cell responses in triply deficient animals (to the point of altering the immunodominance hierarchy) was noteworthy in the face of an immunogen as robust as LCMV.
The defects that we observed in vivo selectively affected the MHC class I antigen processing pathway. Loss of immunoproteasomes weakened the responses to epitopes from several protein antigens but not from a minigene whose product does not require cleavage for presentation. Moreover, we found no defect in CD4+ T cell numbers in response to MHC class II epitopes in vivo, which indicated that APC function was not globally decreased. We also concluded that the weaker CD8+ T cell responses in triply deficient mice were not due to pleiotropic effects on T cells but to antigen-presentation defects, as the differences were found in wild-type T cells transferred into triply deficient hosts. Moreover, we also found no defect in CD4+ T cell numbers or responses in naïve or LCMV-infected animals.
In addition to the differences we observed in the presentation of 9 of 11 immunogenic epitopes, we found that the peptide repertoire of triply deficient animals was qualitatively and substantially different from those of wild-type or any singly deficient mice, as demonstrated by the robust rejection of wild-type cells by triply deficient animals. It should be noted that rejection of the wild-type cells was unlikely to be due to minor histocompatibility differences because the triply deficient animals were fully backcrossed. In addition, such histocompatibility differences would be expected to elicit bidirectional responses between the strains, but we found no rejection of triply deficient cells by wild-type animals. Instead, this 'one-way' rejection suggested that cells in wild-type animals presented a substantially different set of peptides than those found in triply deficient animals, containing epitopes generated by both immunoproteasomes and constitutive proteasomes. Notably, consistent with these results, comparison of the peptides eluted from matched samples of MHC class I molecules on wild-type and triply deficient splenocytes revealed that only about one-half of the peptides from both H-2Db and H-2Kb samples were shared between the two strains. This was probably an underestimate because the mass spectrometry analysis detects the presence of abundant peptides but not their precise amount. Therefore, even among the peptides that were presented in both the wild-type and triply deficient mice, there were probably quantitative differences, as we found such differences in the presentation of the majority (9 of 11) of immunogenic epitopes in quantitative assays. Comparing the peptides we identified as unique to immunoproteasome-deficient mice against a larger data set of BL6-presented peptides from the literature, we found that 75–80% were still present only in the triply deficient pools30. The 20–25% of additional 'shared' peptides could have been generated by constitutive proteasomes in wild-type preparations from the published studies and/or could represent the detection of lower-abundance peptides in published analyses.
Together, these results demonstrated the importance of immunoproteasomes in generating peptides for MHC class I antigen presentation, a contribution that has been substantially underestimated. A potentially important implication of our findings was that under noninflammatory conditions the peptides presented by DCs, which constitutively express immunoproteasomes, will be substantially different from the ones displayed on parenchymal cells, which contain only constitutive proteasomes. Therefore, T cell responses stimulated by DCs may not optimally recognize parenchymal cells until immunoproteasomes are induced in the latter by interferon. This may lower the effectiveness of CD8+ T cell immunity in situations where IFN-γ is not produced. Similarly, this could help pathogenic cells that do not respond to IFN-γ and/or express immunoproteasomes, such as some tumors or cells infected with viruses that inhibit IFN-γ responses, evade immune responses.
Methods
Mice and LCMV.
All mouse strains were maintained in specific pathogen–free conditions in the animal facilities at the University of Massachusetts Medical School (UMMS). All experiments involving live animals were approved by and performed in accordance with guidelines set forth by the UMMS Department of Animal Medicine and the Institutional Animal Care and Use Committee. C57BL/6J, B6.PL-Thy1a/Cy J (Thy1.1), B6;129S2-Tap1tm1Arp/J (Tap1−/−), C57BL/6-Tg(CAG-OVA)916Jen/J (OVA transgenic) were from Jackson Labs. β2i-null (MECL1)16, β1i-null (LMP2)9, β5i-null (LMP7)8 and C57L/J-Tg(Tcrb)93Vbo (H-Y)21,22 mice have been described. Background strain characterization of fully backcrossed triply deficient mice (110 single-nucleotide polymorphisms spanning the genome) was performed by Charles River Laboratories. LCMV-infected mice were housed in the UMMS Biocontainment Suite. Stocks of LCMV, strain Armstrong, were propagated as described31. Mice were infected intraperitoneally with 5 × 104 plaque-forming units of LCMV and were considered immune after at least 6 weeks.
Immunoblotting.
Spleen lymphocytes were lysed in radioimmunoprecipitation buffer with Complete-Mini protease inhibitor cocktail (Roche). Loading of lysates was normalized by protein concentration (Pierce BCA Protein Assay kit). Spleen proteasomes were isolated as described4. Primary antibodies anti-LMP2 (ab3328, Abcam), anti-LMP7 (ab3329, Abcam), anti–α 1+2+3+5+6+7 (MCP231, Abcam), anti-Tap1 (sc-11464, Santa Cruz Biotechnology), anti-GAPDH (MAB374, Millipore) or anti-ubiquitin (Z0458, Dako) were followed with horseradish peroxidase–conjugated secondary antibody (111-035-144 or 115-035-003, Jackson Immunochemicals). Chemiluminescence (Thermo Scientific SuperSignal West Pico or Millipore Immobilon Western HRP Substrates) was detected with X-ray film. The images were optimized with Photoshop Autolevels.
Quantification of Tap1 mRNA.
RNA was harvested with the Qiagen RNeasy kit, treated with Ambion DNA-free reagent (Ambion) and quantified with a Nanodrop spectrophotometer. RNA (1 μg) was then reverse-transcribed with the iScript cDNA synthesis kit (BioRad). TaqMan 2× Master Mix and Tap1 and Actb (β-actin) Primer/Probe sets (ABI) were used for quantitative PCR on a BioRad iCycler.
Flow cytometry analysis of naïve mice.
Lymphocytes (from blood, spleen, thymus or axial, brachial, inguinal and cervical lymph nodes) were blocked with anti–Fc receptor (24G2, ATCC) before being stained with anti-B220 (RA3-6B2, eBioscience), anti-CD3ɛ (145-2C11, BD Pharmingen) or anti–CD3 molecular complex (17A2, BD Pharmingen); anti-CD4 (RM4-5, BD Pharmingen); anti-CD8α (53-6.7, BD Pharmingen); anti-CD11b (M1/70, eBioscience); anti-CD11c (N418, eBioscience), anti–H-2Kb (AF6-88.5, BD Pharmingen) or anti–H-2Db (KH95, BD Pharmingen); and anti-TCRβ (h57-597, eBioscience).
Cell lines.
11p9Z has been described23. For RF33.70-LUC, RF33.70 cells26 were transduced with NFAT-luciferase (from pGL3-NFAT32, Addgene 17870) and the bsdR blastocidin resistance gene (pCDNA6, Invitrogen) driven by the SV40 promoter (pCDNA3.1 hygro+, Invitrogen) cloned into pCDH1 (Systems Bioscience), replacing the CMV promoter (as VSV-G pseudotyped lentivirus). For 12.64-CD8αβ-LUC, 12.64 cells25 were transduced first with pBMN-IRES-Lyt2a (ref. 33; as VSV-G pseudotyped retrovirus, http://www.stanford.edu/group/nolan/), then with pTRIPZ (Open Biosystems) in which the red fluorescent protein had been replaced with CD8b (cloned from mouse spleen cDNA) and finally with pCDH1-NFAT-Luc-Bsd (as lentivirus). Hybridoma culture media was RPMI 1640 with 10% vol/vol FBS, 2 mM L-glutamine, 10 mM HEPES, 1× MEM nonessential amino acids (Gibco) and 55 μM β-mercaptoethanol (Sigma). 12.64-CD8αβ-LUC cells were treated with 1μg/ml doxycycline (Clonetech) for 16–24 h before use.
In vitro antigen presentation.
Bone marrow cells were matured for 6–7 d with granulocyte-macrophage colony-stimulating factor (Invitrogen, 10 ng/ml) and interleukin 4 (IL-4; 5 ng/ml, Invitrogen or eBioscience). For H-Y proliferation, 3 × 104 H-Y CD8α+ cells per well (isolated from the spleen and lymph nodes of female H-Y mice with Miltenyi MACS beads and columns) were cultured for 3 d with BMDCs. [3H]Thymidine (0.5 μCi per well; PerkinElmer) was added, and 3H incorporation was quantified after 5 h. For 11p9Z assay, IL-2 in supernatants from overnight coculture with BMDCs was assayed with CTLL-2 cells34 (Gillis 1977). For influenza assays, 12.64-CD8αβ-LUC cells were cultured overnight with BMDCs infected with influenza strain A/PR/8/34 at 5 × 10−4 hemagglutination units per cell as described35,36. For OVA assays, RF33.70-LUC cells were cultured overnight with irradiated (3,000 rad) splenocytes. For recombinant vaccinia virus assays, RF33.70-LUC were cultured 2–4 h with paraformaldehyde-fixed BMDCs infected with rVV-SIINFEKL37. Luciferase expression was measured with Promega's Luciferase Assay System on a POLARstar OPTIMA luminometer (BMG Labtech).
In vitro stimulation and intracellular cytokine staining.
Spleen leukocytes (2 × 106 to 3 × 106) were stimulated for 5 h at 37 °C with 1 μM of peptides plus 1 μl/ml GolgiPlug (BD Biosciences) and 10 U/ml human recombinant IL-2 (BD Biosciences), as described38. Cells were stained with anti-CD3 (145-2C11, BD Pharmingen); anti-NK1.1 (PK136, BD Pharmingen); anti-CD8a (53-6.7, BD Pharmingen) or anti-CD8b (YTS156.7.7, Biolegend); anti-CD4 (RM4-5, BD Pharmingen); anti-CD44 (IM7, BD Pharmingen); anti-CD62L (MEL-14, BD Pharmingen); anti-Thy1.1 (HIS51, eBioscience); anti-Thy1.2 (53-2.1, BD Pharmingen) and anti-TCR Vα2 (B20.1, BD Pharmingen). These stained cells were fixed and permeabilized with CytoFix/CytoPerm (BD Biosciences), stained with anti–IFN-γ (XMG1.2, eBioscience) and anti-TNF (MP6-XT22, Biolegend) and analyzed on LSRII (BD Biosciences).
In vivo brefeldin A.
Cytokine-producing cells were detected in vivo as described39,40 with modifications. Lymphocytes from spleen and lymph nodes of naïve Thy1.1 P14 TCR-transgenic mice specific for LCMV GP33 were treated with 1 μl/ml GolgiPlug for 15–20 min at 37 °C with shaking every 5 min. Two days after infection, BFA-treated lymphocytes (2 × 107) in 200 μl along with 250 μg BFA in 200 μl were injected intravenously into wild-type and triply deficient mice. Splenocytes were harvested after 4 h for intracellular cytokine staining.
Adoptive transfer.
Wild-type Thy1.1+Thy1.2+ and triply deficient Thy1.1+Thy1.2+ mice were injected with 40 μg anti-Thy1.2 (30H12; BioXCell) per day on days –3, –2, –1 and +4 relative to infection to deplete endogenous T cells. LCMV-immune wild-type Thy1.1 splenocytes (3 × 107) were adoptively transferred intravenously on day –1, and mice were infected intraperitoneally with LCMV on day 0. Splenocytes were harvested 5.5 days after infection for in vitro stimulation and intracellular cytokine staining.
In vivo cytotoxicity.
Spleen lymphocytes from wild-type and immunoproteasome-deficient mice were stained with 1.25 μM (wild-type cells) or 125 nM (immunoproteasome-deficient cells) CFSE (Molecular Probes). Cells were injected intravenously. Percentage wild-type killing is defined as 100 × (1 – (wild-type cells/mutant cells)/(wild-type cells at day 1/mutant cells at day 1)). To deplete host T cells, mice were injected intraperitoneally with 25 μg anti-Thy1.2 on days –2, –1, 0 and +7 relative to splenocyte injection.
Mass spectrometry.
Samples were prepared and analyzed as described41. Manual validation specific to MHC class I peptides was as described42. In the first experiment, 25 wild-type and 50 triply deficient spleens were used. In the second experiment, lysates were normalized for the amount of H-2Kb. The concentration of H-2Kb was measured by ELISA (capture, Y3, ATCC; detection, anti-β2m, A0072, DAKO) and immunoblotting (anti-H-2Kb exon 8; ref. 43). Data from the two experiments were combined in Figure 6.
Statistical analysis.
Statistical analyses were performed with GraphPad Prism version 5.0a for Mac OSX (GraphPad Software).
References
Rock, K.L. et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class. Mol. Cell 78, 761–771 (1994).
Coux, O., Tanaka, K. & Goldberg, A.L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65, 801–847 (1996).
Tanaka, K. Role of proteasomes modified by interferon-gamma in antigen processing. J. Leukoc. Biol. 56, 571–575 (1994).
Gaczynska, M., Rock, K.L. & Goldberg, A.L. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature 365, 264–267 (1993).
Driscoll, J., Brown, M.G., Finley, D. & Monaco, J.J. MHC-linked LMP gene products specifically alter peptidase activities of the proteasome. Nature 365, 262–264 (1993).
Aki, M. et al. Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J. Biochem. 115, 257–269 (1994).
Rammensee, H.G., Falk, K. & Rötzschke, O. Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol. 11, 213–244 (1993).
Fehling, H.J. et al. MHC class I expression in mice lacking the proteasome subunit LMP-7. Science 265, 1234–1237 (1994).
Van Kaer, L. et al. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity 1, 533–541 (1994).
Chen, W., Norbury, C.C., Cho, Y., Yewdell, J.W. & Bennink, J.R. Immunoproteasomes shape immunodominance hierarchies of antiviral CD8+ T cells at the levels of T cell repertoire and presentation of viral antigens. J. Exp. Med. 193, 1319–1326 (2001).
Nussbaum, A.K., Rodriguez-Carreno, M.P., Benning, N., Botten, J. & Whitton, J.L. Immunoproteasome-deficient mice mount largely normal CD8+ T cell responses to lymphocytic choriomeningitis virus infection and DNA vaccination. J. Immunol. 175, 1153–1160 (2005).
Osterloh, P. et al. Proteasomes shape the repertoire of T cells participating in antigen-specific immune responses. Proc. Natl. Acad. Sci. USA 103, 5042–5047 (2006).
Palmowski, M.J. et al. Role of immunoproteasomes in cross-presentation. J. Immunol. 177, 983–990 (2006).
Pang, K.C. et al. Immunoproteasome subunit deficiencies impact differentially on two immunodominant influenza virus-specific CD8+ T cell responses. J. Immunol. 177, 7680–7688 (2006).
Deol, P., Zaiss, D.M.W., Monaco, J.J. & Sijts, A.J.A.M. Rates of processing determine the immunogenicity of immunoproteasome-generated epitopes. J. Immunol. 178, 7557–7562 (2007).
Basler, M., Moebius, J., Elenich, L., Groettrup, M. & Monaco, J.J. An altered T cell repertoire in MECL-1-deficient mice. J. Immunol. 176, 6665–6672 (2006).
Hensley, S.E. et al. Unexpected role for the immunoproteasome subunit LMP2 in antiviral humoral and innate immune responses. J. Immunol. 184, 4115–4122 (2010).
Van Kaer, L. Accessory proteins that control the assembly of MHC molecules with peptides. Immunol. Res. 23, 205–214 (2001).
Macagno, A., Kuehn, L., de Giuli, R. & Groettrup, M. Pronounced up-regulation of the PA28α/β proteasome regulator but little increase in the steady-state content of immunoproteasome during dendritic cell maturation. Eur. J. Immunol. 31, 3271–3280 (2001).
Nil, A., Firat, E., Sobek, V., Eichmann, K. & Niedermann, G. Expression of housekeeping and immunoproteasome subunit genes is differentially regulated in positively and negatively selecting thymic stroma subsets. Eur. J. Immunol. 34, 2681–2689 (2004).
Kisielow, P., Blüthmann, H., Staerz, U.D., Steinmetz, M. & von Boehmer, H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature 333, 742–746 (1988).
Markiewicz, M.A. et al. Long-term T cell memory requires the surface expression of self-peptide/major histocompatibility complex molecules. Proc. Natl. Acad. Sci. USA 95, 3065–3070 (1998).
Sanderson, S. & Shastri, N. LacZ inducible, antigen/MHC-specific T cell hybrids. Int. Immunol. 6, 369–376 (1994).
Greenfield, A. et al. An H-YDb epitope is encoded by a novel mouse Y chromosome gene. Nat. Genet. 14, 474–478 (1996).
Deckhut, A.M. et al. Prominent usage of V beta 8.3 T cells in the H-2Db-restricted response to an influenza A virus nucleoprotein epitope. J. Immunol. 151, 2658–2666 (1993).
Rock, K.L., Rothstein, L. & Gamble, S. Generation of class I MHC-restricted T-T hybridomas. J. Immunol. 145, 804–811 (1990).
Gairin, J.E., Mazarguil, H., Hudrisier, D. & Oldstone, M.B. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J. Virol. 69, 2297–2305 (1995).
Basler, M., Youhnovski, N., Van Den Broek, M., Przybylski, M. & Groettrup, M. Immunoproteasomes down-regulate presentation of a subdominant T cell epitope from lymphocytic choriomeningitis virus. J. Immunol. 173, 3925–3934 (2004).
Stohwasser, R., Standera, S., Peters, I., Kloetzel, P.M. & Groettrup, M. Molecular cloning of the mouse proteasome subunits MC14 and MECL-1: reciprocally regulated tissue expression of interferon-gamma-modulated proteasome subunits. Eur. J. Immunol. 27, 1182–1187 (1997).
Blanchard, N. et al. Endoplasmic reticulum aminopeptidase associated with antigen processing defines the composition and structure of MHC class I peptide repertoire in normal and virus-infected cells. J. Immunol. 184, 3033–3042 (2010).
Welsh, R.M. Jr., Lampert, P.W., Burner, P.A. & Oldstone, M.B. Antibody-complement interactions with purified lymphocytic choriomeningitis virus. Virology 73, 59–71 (1976).
Clipstone, N.A. & Crabtree, G.R. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357, 695–697 (1992).
Levkau, B. et al. xIAP induces cell-cycle arrest and activates nuclear factor-kappaB: new survival pathways disabled by caspase-mediated cleavage during apoptosis of human endothelial cells. Circ. Res. 88, 282–290 (2001).
Gillis, S. & Smith, K.A. Long term culture of tumour-specific cytotoxic T cells. Nature 268, 154–156 (1977).
Sigal, L.J. & Rock, K.L. Bone marrow-derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and -independent pathways of antigen presentation. J. Exp. Med. 192, 1143–1150 (2000).
Oh, S., McCaffery, J.M. & Eichelberger, M.C. Dose-dependent changes in influenza virus-infected dendritic cells result in increased allogeneic T-cell proliferation at low, but not high, doses of virus. J. Virol. 74, 5460–5469 (2000).
Hilton, C.J., Dahl, A.M. & Rock, K.L. Anti-peptide antibody blocks peptide binding to MHC class I molecules in the endoplasmic reticulum. J. Immunol. 166, 3952–3956 (2001).
Brehm, M.A. et al. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat. Immunol. 3, 627–634 (2002).
Liu, F. & Whitton, J.L. Cutting edge: re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J. Immunol. 174, 5936–5940 (2005).
Priyadharshini, B., Welsh, R.M., Greiner, D.L., Gerstein, R.M. & Brehm, M.A. Maturation-dependent licensing of naive T cells for rapid TNF production. PLoS ONE 5, e15038 (2010).
Delgado, J.C., Escobar, H., Crockett, D.K., Reyes-Vargas, E. & Jensen, P.E. Identification of naturally processed ligands in the C57BL/6 mouse using large-scale mass spectrometric peptide sequencing and bioinformatics prediction. Immunogenetics 61, 241–246 (2009).
Escobar, H., Reyes-Vargas, E., Jensen, P.E., Delgado, J.C. & Crockett, D.K. Utility of characteristic QTOF MS/MS fragmentation for MHC class I peptides. J. Proteome Res. 10, 2494–2507 (2011).
Smith, M.H., Parker, J.M., Hodges, R.S. & Barber, B.H. The preparation and characterization of anti-peptide heteroantisera recognizing subregions of the intracytoplasmic domain of class I H-2 antigens. Mol. Immunol. 23, 1077–1092 (1986).
Acknowledgements
We thank J. Monaco (University of Cincinnati) for β2i-deficient mice; L. Van Kaer (Vanderbilt University School of Medicine) for β1i singly deficient mice; C. Perreault (University of Montreal), with permission from H.J. Fehling (University Clinics Ulm) for β5i singly deficient mice; B.J. Fowlkes (US National Institutes of Health) for C57BL/6 H-Y–transgenic mice; N. Shastri (University of California, Berkeley) for11p9z cells; E. Raines (University of Washington) for plasmid pBMN-IRES-Lyt2a; M. Green (University of Massachusetts Medical School) for Phoenix cells and plasmids; Yewdell and Bennick (National Institute of Allergy and Infectious Diseases, US National Institutes of Health) for H-2Kb exon 8–specific antiserum; F. Cruz and D. Farfan (University of Massachusetts Medical School) for the RF33.70-LUC and 12.64-CD8αCD8β-LUC cell lines; D. Palliyaguru for doing many of the in vivo cytotoxicity experiments; and L. Stern and Z. Shen for advice and reagents for optimizing the mass spectrometry. This work was supported by University of Massachusetts Diabetes and Endocrinology Research Center core resources (DK32520; K.L.R., member) and by funding from the US National Institutes of Health (5R01A1020248-29 to K.L.R. and E.Z.K., 5T32A107349-19 and T32CA130807-02 to E.Z.K., R37AI017672 and R01AI081675 to R.M.W. and J.W.C., and U01-AI073871 to J.W.C.).
Author information
Authors and Affiliations
Contributions
E.Z.K. designed and did experiments, analyzed data and wrote the paper; J.W.C. designed and did LCMV experiments and analyzed data; I.Y. designed the β1i β5i doubly deficient mice and did the initial backcrossing; H.E. and E.R.-V. did peptide isolation and mass spectrometry and analyzed data; J.C.D. oversaw the mass spectrometry experiments and data analysis; R.M.W. designed and supervised the LCMV experiments; M.L.K., A.J.M., D.M.V. and G.D.Y. designed the β1i β5i doubly deficient mice; and K.L.R. designed experiments, supervised the experiments and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5 and Supplementary Tables 1–4 (PDF 704 kb)
Rights and permissions
About this article
Cite this article
Kincaid, E., Che, J., York, I. et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol 13, 129–135 (2012). https://doi.org/10.1038/ni.2203
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.2203
This article is cited by
-
Protein degradation by human 20S proteasomes elucidates the interplay between peptide hydrolysis and splicing
Nature Communications (2024)
-
Arg-tRNA synthetase links inflammatory metabolism to RNA splicing and nuclear trafficking via SRRM2
Nature Cell Biology (2023)
-
Interplay between proteasome function and inflammatory responses in e-cig vapor condensate-challenged lung epithelial cells
Archives of Toxicology (2023)
-
A novel and atypical NF-KB pro-inflammatory program regulated by a CamKII-proteasome axis is involved in the early activation of Muller glia by high glucose
Cell & Bioscience (2022)
-
A guide to antigen processing and presentation
Nature Reviews Immunology (2022)
