
Abstract
Congenital generalized lipodystrophy is an autosomal recessive disorder characterized by marked paucity of adipose tissue, extreme insulin resistance, hypertriglyceridemia, hepatic steatosis and early onset of diabetes. We report several different mutations of the gene (AGPAT2) encoding 1-acylglycerol-3-phosphate O-acyltransferase 2 in 20 affected individuals from 11 pedigrees of diverse ethnicities showing linkage to chromosome 9q34. The AGPAT2 enzyme catalyzes the acylation of lysophosphatidic acid to form phosphatidic acid, a key intermediate in the biosynthesis of triacylglycerol and glycerophospholipids. AGPAT2 mRNA is highly expressed in adipose tissue. We conclude that mutations in AGPAT2 may cause congenital generalized lipodystrophy by inhibiting triacylglycerol synthesis and storage in adipocytes.
Similar content being viewed by others
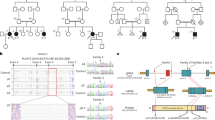

Main
We have previously mapped a form of congenital generalized lipodystrophy (also known as Berardinelli-Seip syndrome) to the CGL1 locus on chromosome 9q34 using genetic linkage analysis of 17 families1. We identified additional families with CGL-affected individuals (CG3700, 5700 and 7000; Fig. 1a), who showed no mutations in BSCL2 (ref. 2), located on chromosome 11q13. Linkage to CGL1 was confirmed in these pedigrees using 9q34 microsatellite markers as previously described1. Recombination mapping in pedigrees CG7000 and CG600 reduced the critical region to approximately 0.86 Mb between rs1747838 and DPP7 (see Fig. 1b, and Web Fig. A and Web Table A online). Sequencing of the exons and splice-site junctions of AGPAT2 located in this interval3,4 revealed homozygous or compound heterozygous mutations in CGL-affected individuals of 11 pedigrees showing linkage to 9q34 (see Table 1 and Web Fig. A online).

a, Pedigrees of families with CGL. Individuals for whom DNA was available are indicated by an arrowhead. For simplicity, only those individuals who contribute to understanding of the consanguinity loops are shown for pedigree CG7000. b, Haplotypes of affected individuals from the CG7000 pedigree, linked to the 9q34 region. Gray areas indicate regions of homozygosity. Microsatellite loci are shown on the left. The individuals are numbered as shown in a. The paternal haplotype is shown in the right bar and the maternal haplotype in the left bar. c, Mutations in individuals of pedigree CG7000. Deletion of exons 3 and 4 is seen in affected individuals from CG7000, whereas parents of affected subjects show no deletions. d, Expression of AGPAT mRNAs in human tissues.Gel images obtained from RT–PCR show that in adipose tissue, AGPAT2 was expressed twofold more than AGPAT1. In liver, expression of both AGPAT1 and AGPAT2 was the same; in skeletal muscle, AGPAT1 was expressed 1.8-fold more than AGPAT2. Expression of AGPAT3, AGPAT4 and AGPAT5 was barely detectable. The results were normalized to the signal generated from G3PDH. Details of the methods are provided as Web Note A.
Affected individuals from pedigree CG7000 were homozygous with respect to a deletion of exons 3 and 4 (Fig. 1c) resulting in a frameshift mutation and premature termination codon. This mutation was confirmed in lymphoblast-derived cDNA of individual CG7000.8. Affected individuals homozygous for an identical nonsense mutation (Arg63X) were identified in families from Turkey (CG5700) and Belgium (CG3700). A homozygous splice-site mutation resulting in a deletion of two amino acids starting at codon 221 was found in the affected individual from pedigree CG3500. Affected individuals from two African American families (CG400 and CG900) were homozygous with respect to a splice-site mutation (IVS4-2A→G) resulting in a frameshift and a premature termination codon (Gln196fsX228). This same splice-site mutation was identified in the heterozygous state in individuals from pedigrees CG3200, CG600 (African Caribbean families) and CG800 (African American family). Affected compound heterozygotes from these families were found to have a missense mutation, a frameshift mutation and a single–amino acid deletion, respectively. The affected individual in pedigree CG700 was a compound heterozygote having one missense and one frameshift mutation. The affected individual from pedigree CG3300 was found to be heterozygous for a missense mutation. We did not detect a coding or splice-site mutation on the other allele; however, we did find a mutation in the 3′ UTR, which we suggest has a deleterious effect on gene expression.
Mutations segregated in all pedigrees in accordance with autosomal recessive inheritance. Sequencing of exons 2–5, which contained all of the mutations seen in individuals with CGL, of 50 unaffected subjects (25 each of European and African origin) revealed no mutations. All of the chromosomes carrying the IVS4-2A→G mutation in five families of African origin had the same haplotype for seven markers extending 33 kb, suggesting a common ancestral mutation (see Web Table B online).
The AGPAT2 protein has 278 amino acids and belongs to the family of acyltransferases5 and catalyzes an essential reaction in the biosynthetic pathway of glycerophospholipids and triacylglycerol in eukaryotes. To our knowledge, CGL is the first documented human disease caused by a mutation in this pathway. The first step of the pathway involves glycerol-3-phosphate acyltransferase (GPAT), which acylates glycerol-3-phosphate at the first carbon (sn-1 position) to form lysophosphatidic acid. AGPAT2 catalyzes acylation of lysophosphatidic acid at the second carbon (sn-2 position) to form phosphatidic acid. Dephosphorylation of phosphatidic acid occurs next, to form 1,2 diacylglycerol, which is acylated to form triacylglycerol using diacylglycerol acyltransferase (DGAT).
The AGPAT2 protein shows 48% identity with AGPAT1, and much less homology to three other related proteins AGPAT3, AGPAT4 and AGPAT5 (refs 5,7). All known AGPATs share two motifs: NHX4D, which is involved in catalytic function, and EGTR, which is involved in substrate binding and recognition. The mutations Arg68X, Gly106fsX188 and Leu126fsX146 affect either one or both motifs. The two splice-site mutations retain these motifs; however, IVS4-2A→G results in an aberrant and truncated protein and IVS5-2A→C causes the deletion of two amino acids and may disrupt secondary structure of the protein, as can the mutation 140delPhe. The three missense mutations, Leu228Pro, Ala239Val and Gly136Arg, result in nonconservative amino-acid changes at evolutionary conserved sites between the human and mouse proteins.
The AGPAT1 mRNA is ubiquitously expressed in human tissues, with the greatest amount of expression in skeletal muscle6. Expression of AGPAT2 is more tissue-restricted, occuring at high levels in liver and heart tissues but almost undetectable in brain3,4. Amplification of all AGPAT mRNA in normal human omental adipose tissue revealed at least twofold higher expression of AGPAT2than AGPAT1 (Fig. 1d). AGPAT2 was expressed at lower level in liver and much less in skeletal muscle. Homologues of AGPAT2, AGPAT3, AGPAT4 and AGPAT5 were barely detectable (Fig. 1d).
The AGPAT2 enzyme is likely to affect triacylglycerol synthesis in adipose tissue and may cause lipodystrophy by resulting in triglyceride-depleted adipocytes. It is also likely that reduced AGPAT2 activity could increase tissue levels of lysophosphatidic acid, which may affect adipocyte functions8. Lysophosphatidic acid is a ligand for G-protein coupled receptors and may have a role in preadipocyte proliferation and adipogenesis8,9. Decreased AGPAT2 activity could also lead to reduced bioavailability of phosphatidic acid and phosphoglycerols (phosphatidylinositol, phosphatidylcholine and phosphatidylethanolamine), which are important in intracellular signaling and could affect adipocyte functions10.
The gene BSCL2 encodes a protein of unknown function2. Individuals with CGL who carry BSCL2 mutations seem to have mild mental retardation and cardiomyopathy11,12, which are not seen in the individuals we studied who have AGPAT2 mutations. Based on the high expression of BSCL2 in brain and weak expression in adipocytes, a primary defect in hypothalamic pituitary axis has been suggested2. Our data suggest that mutations in AGPAT2 may cause congenital generalized lipodystrophy by inhibiting triacylglycerol synthesis in adipocytes. Thus, different forms of congenital generalized lipodystrophy may be caused by disruption of distinct pathways.
GenBank accession numbers.
AGPAT1 cDNA, XM_057885; AGPAT2 cDNA, AF000237; AGPAT3 cDNA, XM_036003; AGPAT4 cDNA, XM_004374; AGPAT5 cDNA, XM_034829; mouse Agpat2 cDNA, AY072769.
Note: Supplementary information is available on the Nature Genetics website.
References
Garg, A. et al. 84, 3390–3394 (1999).
Magre, J. et al. 28, 365–370 (2001).
Eberhardt, C., Gray, P.W. & Tjoelker, L.W. J. Biol. Chem. 272, 20299–20305 (1997).
Eberhardt, C., Gray, P.W. & Tjoelker, L.W. Adv. Exper. Med. Biol. 469, 351–356 (1999).
Leung, D.W. Fron. in Biosci. 6, D\944–953 (2001).
West, J. et al. DNA Cell. Biol. 16, 691–701 (1997).
Lewin, T.M., Wang, P. & Coleman, R.A. Biochemistry 38, 5764–5771 (1999).
Pages, C., Simon, M., Valet, P. & Saulnier-Blache, J.S. Prostaglandins Other Lipid Mediat. 64, 1–10 (2001).
Hla, T., Lee, M.J., Ancellin, N., Paik, J.H. & Kluk, M.J. Science 294, 1875–1878 (2001).
Fang, Y., Vilella-Bach, M., Bachmann, R., Flanigan, A. & Chen, J. Science 294, 1942–1945 (2001).
Vigouroux, C. et al. J. Clin. Endocrinol. Metab. 82, 3438–3444 (1997).
Seip, M. & Trygstad, O. Acta Pædiatrica 413 (suppl.), 2–28 (1996).
Huseman, C., Johanson, A., Varma, M. & Blizzard, R.M. J. Pediatr. 93, 221–226 (1978).
Garg, A., Fleckenstein, J.L., Peshock, R.M. & Grundy, S.M. J. Clin. Endocrinol. Metab. 75, 358–361 (1992).
Oral, E.A. et al. N. Engl. J. Med. 346, 570–578 (2002).
Acknowledgements
We thank the members of the families studied for their invaluable contribution to this project; A. Osborn, T. Petricek and A. Nguyen for management of DNA and patient databases, illustrations and technical assistance; R. Wilson and B. Crider for help in genotyping; J. Cohen for providing DNA samples of control subjects; C. Helms and J. Dawes for technical assistance and the nursing and dietetic services of the General Clinical Research Center for patient care support. This work was supported by grants from the National Institutes of Health and by the Southwestern Medical Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Agarwal, A., Arioglu, E., de Almeida, S. et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet 31, 21–23 (2002). https://doi.org/10.1038/ng880
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng880
This article is cited by
-
Phospholipid biosynthetic pathways and lipodystrophies: a novel syndrome due to PLAAT3 deficiency
Nature Reviews Endocrinology (2024)
-
Lipid droplet biogenesis and functions in health and disease
Nature Reviews Endocrinology (2023)
-
Dynamic chromatin architecture of the porcine adipose tissues with weight gain and loss
Nature Communications (2023)
-
Deficiency of WTAP in hepatocytes induces lipoatrophy and non-alcoholic steatohepatitis (NASH)
Nature Communications (2022)
-
Lipodystrophy for the Diabetologist—What to Look For
Current Diabetes Reports (2022)
