
Abstract
Hermansky–Pudlak syndrome (HPS) is a rare autosomal recessive disorder characterized by oculocutaneous albinism and a storage pool deficiency due to an absence of platelet dense bodies1,2,3. Lysosomal ceroid lipofuscinosis, pulmonary fibrosis and granulomatous colitis are occasional manifestations of the disease4. HPS occurs with a frequency of one in 1,800 in north-west Puerto Rico5 due to a founder effect6. Several non-Puerto Rican patients also have mutations in HPS17,8, which produces a protein of unknown function9. Another gene, ADTB3A, causes HPS in the pearl mouse10 and in two brothers with HPS-2 (refs. 11,12). ADTB3A encodes a coat protein involved in vesicle formation3,13, implicating HPS as a disorder of membrane trafficking. We sought to identify other HPS-causing genes7,8,14. Using homozygosity mapping on pooled DNA of 6 families from central Puerto Rico, we localized a new HPS susceptibility gene to a 1.6-cM interval on chromosome 3q24. The gene, HPS3, has 17 exons, and a putative 113.7-kD product expected to reveal how new vesicles form in specialized cells. The homozygous, disease-causing mutation is a large deletion and represents the second example of a founder mutation causing HPS on the small island of Puerto Rico. We also present an allele-specific assay for diagnosing individuals heterozygous or homozygous for this mutation.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
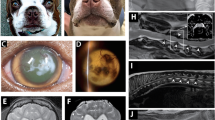

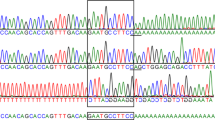
References
Hermansky, F. & Pudlak, P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: report of two cases with histochemical studies. Blood 14, 162–169 (1959).
King, R.A., Hearing, V.J., Creel, D.J. & Oetting, W.S. in The Metabolic and Molecular Bases of Inherited Disease 8th edn., Vol. IV (eds. Scriver, C.R., Beaudet, A.L., Sly, W.S. & Valle, D.L.) 5587–5627 (McGraw-Hill, New York, 2001).
Huizing, M., Anikster, Y. & Gahl, W.A. Hermansky–Pudlak syndrome and related disorders of organelle formation. Traffic 1, 823–835 (2000).
Gahl, W.A. et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky–Pudlak syndrome). N. Engl. J. Med. 338, 1258–1264 (1998).
Witkop, C.J. et al. Albinism and Hermansky–Pudlak syndrome in Puerto Rico. Bol. Asoc. Med. P. R.. 82, 333–339 (1990).
Oh, J. et al. Positional cloning of a gene for Hermansky–Pudlak syndrome, a disorder of cytoplasmic organelles. Nature Genet. 14, 300–306 (1996).
Shotelersuk, V. & Gahl, W.A. Hermansky–Pudlak syndrome: models for intracellular vesicle formation. Mol. Genet. Metab. 65, 85–96 (1998).
Oh, J. et al. Mutation analysis of patients with Hermansky–Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity. Am. J. Hum. Genet. 62, 593–598 (1998).
Dell'Angelica, E. et al. Molecular characterization of the protein encoded by the Hermansky–Pudlak syndrome type 1 gene. J. Biol. Chem. 275, 1300–1306 (2000).
Feng, L. et al. The beta3A subunit gene (Ap3b1) of the AP-3 adaptor complex is altered in the mouse hypopigmentation mutant pearl, a model for Hermansky–Pudlak syndrome and night blindness. Hum. Mol. Genet. 8, 323–330 (1999).
Dell'Angelica, E.C., Shotelersuk, V., Aguilar, R.C., Gahl, W.A. & Bonifacino, J.S. Altered trafficking of lysosomal proteins in Hermansky–Pudlak syndrome due to mutations in the beta3A subunit of the AP-3 adaptor. Mol. Cell. 3, 11–21 (1999).
Shotelersuk, V., Dell'Angelica, E.C., Hartnell, L., Bonifacino, J.S. & Gahl, W.A. A new variant of Hermansky–Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am. J. Med. 108, 423–427 (2000).
Dell'Angelica, E.C., Ooi, C.E. & Bonifacino, J.S. Beta3A-adaptin, a subunit of the adaptor-like complex AP-3. J. Biol. Chem. 272, 15078–15084 (1997).
Hazelwood, S. et al. Evidence for locus heterogeneity in Puerto Ricans with Hermansky–Pudlak syndrome. Am. J. Hum. Genet. 61, 1088–1094 (1997).
Witkop, C.J., Krumwiede, M., Sedano, H. & White, J.G. Reliability of absent platelet dense bodies as a diagnostic criterion for Hermansky–Pudlak syndrome. Am. J. Hematol. 26, 305–311 (1987).
Swank, R.T., Novak, E.K., McGarry, M.P., Rusiniak, M.E. & Feng, L. Mouse models of Hermansky Pudlak syndrome: a review. Pigment Cell Res. 11, 60–80 (1998).
Swank, R.T., Reddington, M. & Novak, E.K. Inherited prolonged bleeding time and platelet storage pool deficiency in the subtle gray (sut) mouse. Lab. Anim. Sci. 46, 56–60 (1996).
Emr, S.C. & Malhota, V.V. Membranes and sorting. Curr. Opin. Cell Biol. 9, 475–476 (1997).
Dell'Angelica, E.C., Klumperman, J., Stoorvogel, W. & Bonifacino, J.S. Association of the AP-3 adaptor complex with clathrin. Science 280, 431–434 (1998).
Höning, S., Sandoval, I.V. & von Figura, K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosine mediates selective binding of AP-3. EMBO J. 17, 1304–1314 (1998).
Ohno, H., Fournier, M.C., Poy, G. & Bonifacino, J.S. Structural determinants of interaction of tyrosine-based sorting signals with the adaptor medium chains. J. Biol. Chem. 271, 29009–29015 (1996).
Teasdale, R.D. & Jackson, M.R. Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu. Rev. Cell. Dev. Biol. 12, 27–54 (1996).
McNew, J.A. & Goodman, J.M. The targeting and assembly of peroxisomal proteins: some old rules do not apply. Trends Biochem. Sci. 21, 54–58 (1996).
Toro, J., Turner, M. & Gahl, W.A. Dermatologic manifestations of Hermansky–Pudlak syndrome in patients with and without a 16-base pair duplication in the HPS1 gene. Arch. Dermatol. 135, 774–780 (1999).
Iwata, F. et al. Correlation of visual acuity and ocular pigmentation with the 16-bp duplication in the HPS-1 gene of Hermansky–Pudlak syndrome, a form of albinism. Ophthalmology 107, 783–789 (2000).
Luria, S.E. & Delbruck, M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28, 491–511 (1943).
Santiago-Gonzalez, J. & Cardona-Bonet, W.A. Aibonito notas para su historia (Oficina estatal de preservacion historica, San Juan, Puerto Rico, 1985).
Antonarakis, S.E., Krawczak, M. & Cooper, D.N. in The Metabolic and Molecular Bases of Inherited Disease 8th edn, Vol. I (eds. Scriver, C.R., Beaudet, A.L., Sly, W.S. & Valle, D.L.) 359 (McGraw-Hill, New York, 2001).
Deininger, P.L. & Batzer, M.A. Alu repeats and human disease. Mol. Genet. Metab. 67, 183–193 (1999).
Sambrook, J., Fritsch, E.F. & Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd edn (Cold Spring Harbor, NY, Cold Spring Harbor Laboratory Press, 1989).
Acknowledgements
Y. Anikster is a Howard Hughes Medical Institute Physician Postdoctoral Fellow. The authors thank J.Sanchez, Chairman, Department of Dermatology, University of Puerto Rico-Medical Sciences Campus, for his helpful assistance, J.L. Arribas for his genealogical information and C.N. Rodriguez for serving as patient liaison. The authors appreciate the advice of R. King of the University of Minnesota, the consultation of R. Kleta and the excellent technical assistance of I. Bernardini. Supported in part by grant HL31698 (RTS) and by the Roswell Park Cancer Institute Cancer Center Support Grant CA16506.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Rights and permissions
About this article
Cite this article
Anikster, Y., Huizing, M., White, J. et al. Mutation of a new gene causes a unique form of Hermansky–Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet 28, 376–380 (2001). https://doi.org/10.1038/ng576
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng576
This article is cited by
-
Hermansky-Pudlak syndrome and oculocutaneous albinism in Chinese children with pigmentation defects and easy bruising
Orphanet Journal of Rare Diseases (2019)
-
Matrix metalloproteinase activity in the lung is increased in Hermansky-Pudlak syndrome
Orphanet Journal of Rare Diseases (2019)
-
Novel HPS6 mutations identified by whole-exome sequencing in two Japanese sisters with suspected ocular albinism
Journal of Human Genetics (2016)
-
Homozygosity Mapping and Whole-Exome Sequencing to Detect SLC45A2 and G6PC3 Mutations in a Single Patient with Oculocutaneous Albinism and Neutropenia
Journal of Investigative Dermatology (2011)
-
Clinical, Molecular, and Cellular Features of Non-Puerto Rican Hermansky–Pudlak Syndrome Patients of Hispanic Descent
Journal of Investigative Dermatology (2011)
