
Abstract
PDGF-C is a member of the platelet-derived growth factor (PDGF) family, which signals through PDGF receptor (PDGFR) αα and αβ dimers1,2. Here we show that Pdgfc−/− mice die in the perinatal period owing to feeding and respiratory difficulties associated with a complete cleft of the secondary palate. This phenotype was less severe than that of Pdgfra−/− embryos. Pdgfc−/− Pdgfa−/− embryos developed a cleft face, subepidermal blistering, deficiency of renal cortex mesenchyme, spina bifida and skeletal and vascular defects. Complete loss of function of both ligands, therefore, phenocopied the loss of PDGFR-α function, suggesting that both PDGF-A and PDGF-C signal through PDGFR-α to regulate the development of craniofacial structures, the neural tube and mesodermal organs. Our results also show that PDGF-C signaling is a new pathway in palatogenesis, different from, and independent of, those previously implicated.
Similar content being viewed by others

Main
To investigate the role of PDGF-C in development, we studied the effect of inactivating Pdgfc by gene-targeting in mice (Fig. 1a). We confirmed correct homologous recombination in R1 embryonic stem (ES) cells by Southern-blot analysis (Fig. 1b). RNA-blot hybridization (Fig. 1c) showed that Pdgfc mRNA was absent in the homozygous embryos, indicating that the mutation had generated a null allele. In Pdgfc+/− embryos, lacZ reporter activity recapitulated the endogenous Pdgfc expression pattern (Fig. 1d). Among the offspring of intercrosses of Pdgfc+/− (129S1) mice, Pdgfc−/− fetuses were present in a mendelian ratio up to birth, suggesting that Pdgfc deficiency does not cause prenatal lethality. But more than 95% of Pdgfc−/− neonates (83 of 87) died on day 1, and none survived to weaning.

(a) Structure of the wild-type Pdgfc locus, the targeting vector and the mutated locus with exon 2 replaced by the SA-IRES-βgeopA cassette. Two restriction digestions were done: SacI digestion and probing with 5′ probe produced a 7-kb wild-type allele and a 9-kb targeted allele; BamH1 digestion and probing with a 3′ probe produced a 3.6-kb wild-type band and a 4.7-kb targeted band. (b) Southern-blot analysis of targeted DNA from ES cell clones digested with SacI and probed with a 5′ probe. (c) RNA-blot analysis of total RNA from wild-type (+/+) and Pdgfc−/− embryos (−/−; at 10.5 d.p.c.). (d) X-gal staining of a 9.5-d.p.c. Pdgfc+/− embryo.
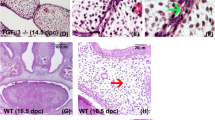
In the Pdgfc−/− embryos, we detected no morphological defect in tissues that normally express Pdgfc, but the null mutant embryos had subcutaneous edema in the flank of the body between the limbs. We observed severe blistering in the frontonasal and lateral forehead region, and blisters were often filled with blood owing to the rupture of extracranial vessels (Fig. 2). We observed subepithelial blistering in the posterior part of the secondary palate and the nasal septum (Fig. 3m). Ultrastructural analysis showed that the roof of the blister was lined by intact lamina lucida and lamina densa, but very few anchoring fibrils were found underneath the lamina densa. This indicated that the cleavage plane lies in the sublamina densa zone on the dermal side of the basement membrane (Fig. 2e,f). The histopathology of the subepidermal blistering in the mutant embryo was reminiscent of that of dystrophic epidermolysis bullosa in humans.

(a) Subepidermal blood-filled blisters in the frontal (FR) and frontonasal (FN) regions of a 12.5-d.p.c Pdgfc−/− embryo. (b) The hematoma (HM) between the epidermis and the dermal tissues of the frontonasal region of a 15.5-d.p.c. Pdgfc−/− embryo. FC, frontal cortex; NS, nasal septum. (c,d) Blisters in the dorsal side of the spinal cord (c) and the dorsal side of the limb (d) of 15.5 d.p.c Pdgfc−/− embryos. BL, subepidermal blister; PL, phalangeal cartilage; SK, skin; SC, spinal cord; TM, tendon and muscles; VB, truncated neural arch of the vertebra. (e,f) The dermo-epidermal junction of a wild-type embryo (e) and of the blister of a 13.5-d.p.c. Pdgfc−/− embryo (f). Arrows indicate the lamina lucida (LL), lamina densa (LD) and anchoring fibrils (AF) beneath the epithelium. (× 50,000). N, nucleus.

(a) The cleft palate (arrow) of a Pdgfc−/− neonate. (b) The palatal bones in the Pdgfc−/− mutant failed to meet (arrows; compare with wild-type embryo, c). (d) Spina bifida occulta of a 15-d.p.c. Pdgfc−/− embryo (arrow indicates a blister). (e) Thoracic vertebrae of wild-type (WT) and Pdgfc−/− (−/−) neonates. (f–m) Palatogenesis in coronal sections (stained with hematoxylin and eosin) of Pdgfc+/− (f,h,j) and Pdgfc−/− (g,i,k,l,m) embryos at 13.5 d.p.c. (f,g), 14.5 d.p.c. (h,i) and 14.75 d.p.c. (j–m). (n,o) The Ki67-positive bulging cells (arrows) of the palatal shelves (PS) of wild-type (n) and Pdgfc−/− (o) embryos. (p,q) Filopodia are absent in the MEE cells of a Pdgfc−/− embryo (q; compare with wild-type embryo, p; × 6,400). (r–bb) Expression of Pdgfc (r), Pdgfa (s,t), Pdgfra (u,v), Msx1(w,x), Irf6 (y,z) and Pvrl1 (aa,bb) in palatal shelves (PS) of wild-type (r,s,u,w,y,aa) and Pdgfc−/− (t,v,x,z,bb) embryos. (cc–dd) Pdgfc−/− expression in palatal shelves (PS) of wild-type (cc) and Tgfb3−/− (dd) embryos. (ee) TGFβ3 immunostaining of the palatal shelf of a Pdgfc−/− embryo. Embryos in r–ee are 13.5 d.p.c. T, tongue.
All Pdgfc−/− neonates that died shortly after birth showed a complete cleft of the secondary palate (Fig. 3a), accompanied by the failure of the palatal bones to extend across the roof of the oronasal cavity (Fig. 3b; compare with wild-type control in Fig. 3c). We observed no other skull abnormalities (data not shown), but spina bifida occulta (incomplete neural arch of the vertebra and deformation of the dorsal spinal cord) developed in the lower spine (Fig. 3e). This might be associated with the formation of subepidermal blisters and the deficiency of connective tissues in the dorsal midline (Fig. 3d).
We investigated the dysmorphogenesis of the palatal shelf of Pdgfc mutant embryos. At 13.5 days post-coitum (d.p.c.), the palatal shelves of both Pdgfc+/− and Pdgfc−/− embryos showed a proper downward growth from the maxillary processes (Fig. 3f,g). By 14.5 d.p.c., the palatal shelves of Pdgfc+/− embryos were elevated to a horizontal position and abutted each other across the gap dorsal to the tongue, which had been lowered from between the shelves (Fig. 3h,i). In contrast, the palatal shelves of the Pdgfc−/− embryos (n = 9) failed to lift, even after the tongue had moved away from between them. Both shelves and the nasal septum were short for the size of the gap in the oro-pharynx (Fig. 3i). In Pdgfc+/− embryos at 14.75–15.5 d.p.c., the medial-edge epithelia (MEE) of the two palatal shelves fused and then degenerated, resulting in congruity of the epithelial and mesenchymal tissues of the shelves (Fig. 3j). In contrast, the palatal shelves of all five Pdgfc−/− embryos at similar ages were hypoplastic and failed to fuse properly (Fig. 3k–m). Four of them had asymmetrical fusion of the nasal septum (Fig. 3l) and three had blisters on the MEE region (Fig. 3m).
The proliferation of bulging MEE cells and the modification of the surface of MEE, such as the formation of filopodia on the apical surfaces, are indicative of the adhesive property of the palatal shelf (Fig. 3n,p)3,4. In the palatal shelf of Pdgfc−/− embryos, the number of proliferating bulging MEE cells was reduced markedly, and the mesenchyme immediately subjacent to the MEE was depleted of cells (Fig. 3o). Filopodia were lost completely from the surface of MEE cells (Fig. 3q). During the closure of dorsal epithelial folds in Drosophila melanogaster and in ventral epithelial folds in Caenorhabditis elegans, the presence of filopodia enables the epithelial cell to sense and establish contact with other epithelial cells. In concert with the E-cadherin that clusters at the tips of filopodia, adherens junctions are formed between cells5,6. The MEE filopodia might have a similar function during palate fusion, drawing cells together to facilitate recognition and adhesion between the apposing MEE. The absence of the MEE filopodia might have contributed to the poor ability of the palatal shelves of 13.5-d.p.c. Pdgfc−/− embryos to fuse, even when they were apposed closely in vitro (Supplementary Table 1 online). Together, the hypoplasia of palate tissues and fusion defects of the Pdgfc−/− MEE might account for the formation of a cleft palate in Pdgfc−/− neonates.
Both Pdgfc (Fig. 3r) and Pdgfa (Fig. 3s), which encode PDGF ligands, were expressed in the epithelium of the palatal shelf, whereas Pdgfra, which encodes the receptor, was expressed in the mesenchyme (Fig. 3u). In palatal shelves of Pdgfc−/− mutants, Pdgfa (Fig. 3t) and Pdgfra (Fig. 3v) were still expressed, although the latter might have been slightly downregulated. The regional activity of the ligand and receptor is suggestive of a paracrine mode of signaling in vivo7,8. The MEE cell phenotype might therefore be secondary to the effect of diminishing PDGF signaling in the mesenchyme owing to the loss of the epithelial source of PDGF-C (Fig. 3o). PDGF-C function is probably nonredundant in the formation of the secondary palate, as the palate defects were not rescued by the Pdgfa activity in the MEE, and the loss of PDGF-A did not cause a cleft palate in Pdgfa−/− mice7.
Pdgfra−/− mutants die by 15.5 d.p.c. and have a severe phenotype of cleft face, spina bifida, subepidermal blistering and skeletal and vascular defects9,10. Although most Pdgfa−/− mutants die during embryonic development, some are born with abnormalities of the central nervous system11, the skin12, the lung13 and the gut14, but without the facial cleft that is characteristic of Pdgfra mutants. As expression of Pdgfc largely overlaps with that of Pdgfa during early embryogenesis15,16, the loss of PDGFR-α function may result from the combined effects of loss of the activity of both ligands. To test this idea, we crossed Pdgfc+/− mice (129S1) with Pdgfa+/− mice (C57/B6) to generate trans-heterozygous mice, which we then bred to produce Pdgfc−/− Pdgfa−/− mice. We recovered the expected numbers (1 of 16) of Pdgfc−/− Pdgfa−/− embryos at 8.5–10.5 d.p.c. but found fewer at later embryonic ages (Table 1). Like Pdgfra−/− mutants, most 10.5-d.p.c. Pdgfc−/− Pdgfa−/− homozygotes (16 of 20) showed growth retardation, pericardial effusion (Fig. 4a), wavy neural tube (Fig. 4e) and subepidermal blisters (Fig. 4b). Double homozygotes (n = 4) were grossly normal (Fig. 4a), but the myotome of somites in the lower occipital and cervical region was necrotic and poorly organized (Fig. 4n,o). To procure more compound homozygous embryos for phenotype analysis, we produced Pdgfc−/− Pdgfa−/− embryos totally from ES cells using tetraploid chimera technology17. We obtained 112 ES cell–derived Pdgfc−/− Pdgfa−/− embryos, which were phenotypically similar to the compound homozygotes from Pdgfc+/− Pdgfa+/− intercrosses (data not shown). The 10.5-d.p.c ES cell–derived Pdgfc−/− Pdgfa−/− embryos showed disrupted metameric organization of the myotomes (Pdgfc, Fig. 4c,d; and Pax3, Fig. 4k–m), the dermomyotome (En1, Fig. 4f,g) and the sclerotome (Twist1, Fig. 4h–j) of somites in the cervical region; somites in the trunk were unaffected. Older Pdgfc−/− Pdgfa−/− embryos developed a cleft face (Fig. 5b,d,g; compare with wild-type controls in Fig. 5a,c), edema and subepidermal blistering (Fig. 5e,f,h), and skeletal defects including cranial bone defects (data not shown), fusion or bifurcation of ribs, short sternum (Fig. 5j), retarded formation of the acromion of the scapula (Fig. 5k) and incomplete neural arch of vertebrae (resulting in spina bifida; Fig. 5i,l). The hearts of the Pdgfc−/− Pdgfa−/− mutants lacked atrial septa (Fig. 5m), and the kidney cortex was deficient in interstitial mesenchyme (Fig. 5o; compare with wild-type control in Fig. 5n). All of these phenotypes were present in Pdgfra−/− mutant embryos9,10.

(a) The morphology of the mutants varied from normal (left) to severely malformed (right; arrow indicates enlarged pericardium). (b) Subepidermal blisters (arrows). (c) Disorganized myotome (arrows) in the cervical somites of a Pdgfc−/− Pdgfa−/− embryo (at 10.5 d.p.c.) compared with a wild-type embryo (d). (e) Abnormal cervical neural tube of the Pdgfc−/− Pdgfa−/− embryo. (f–m) Expression of En1 (f,g), Twist1 (h–j) and Pax3 (k–m) in wild-type (f,h,k) and Pdgfc−/− Pdgfa−/− (g,i,j,l,m) embryos showing disorganized meristic pattern (g,j,m), missing sclerotomes (i), reduced myotome (l) and missing myotome (m). (n,o) Degeneration of myotome (n; arrow) and somite-derived mesenchyme (o) around the dorsal root ganglion in the cervical region.

(a–d) Widened gap between the nasal folds at 11.0 d.p.c. (b) and failure of fusion of nasal processes at 11.5 d.p.c. (d; compare with wild-type embryo, a,c). (e–g) Formation of blisters (e; 11.0 d.p.c.) and partial (f; 11.25 d.p.c.) and complete (g) loss of frontonasal tissues (arrow, membraneous remnant; 11.5 d.p.c.). (h,i) Cleft face, subepidermal blistering (h; lateral view) and spina bifida (i; dorsal view; 13.5 d.p.c.). (j–l) Rib bifurcation (j) and cervical vertebral fusion (j), poor development of the acromion of the scapula (k) and open neural arches (l; both 14.5 d.p.c.). (m) Loss of atrial septa and gap in the interventicular septum (15.5 d.p.c.). (n,o) Deficiency of renal mesenchyme (arrow; 15.5 d.p.c.). n, wild-type; o, mutant.
Our results show that PDGF-C is a key component of the PDGFR-α signaling pathway and has a specific role in palatogenesis and the morphogenesis of the integumentary tissue. The phenotypes of compound mutants imply that PDGF-C and PDGF-A may function as principal ligands for PDGFR-α. Although another ligand, PDGF-B, can bind to PDGFR-α with high affinity, loss of its function18 produces a phenotype that is substantially different from that resulting from loss of PDGF-A, PDGF-C and PDGFR-α. These findings suggest that PDGF-B does not function through PDGFR-α signaling and therefore probably does not contribute to the phenotype resulting from loss of PDGF-A and PDGF-C. The role of PDGF-D, however, has not been investigated. Loss of function of another receptor (PDGFR-β) is associated with a different phenotype19, and loss of function of both PDGF-A and PDGF-B does not result in any of the phenotypes associated with mutations of PDGFR-α (C.B and H.B., unpublished observations). These results suggest that the function of the two receptors is not redundant.
Clefts of the lip or palate (CLP) are complex human congenital anomalies20. Despite intensive efforts in mutation screening and animal studies, the elucidation of the genetic cause is far from complete. Nevertheless, significant association has been found between human nonsyndromic CLP and nonsense mutations of several genes, such as PVRL1 (ref. 21), IRF6 (ref. 22) and MSX1 (ref. 23). Our results show that expression of the mouse homologs of these genes was unaltered in Pdgfc−/− mutant embryos (Fig. 3w–bb), suggesting that their activity is not related to PDGF-C signaling in palatogenesis. As failure of secondary palate fusion is associated with Tgfβ3 deficiency24,25, we examined Pdgfc in Tgfb3−/− embryos but found it to be normal. (Fig. 3dd) Conversely, Tgfb3 expression was unchanged in Pdgfc−/− embryos (Fig. 3ee). These results confirm that PDGF-C signaling is a new and independent mechanism that regulates palatogenesis. PDGF-C is a latent growth factor with proteolytic activation1, and the processing enzyme might be controlled by the other CLP-associated genes that may indirectly connect to PDGF-C signaling. Notably, a 30-cM region on human chromosome 4, where the PDGFC gene maps, shows strong linkage association with CLP26, and our unpublished clinical genetic data further suggest a potential link between PDGFC gene polymorphism and CLP.
Methods
Construction of targeting vector and derivation of mice deficient for PDGF-C.
We cloned mouse Pdgfc genomic DNA from a 129S1 genomic library. We used a ∼12-kb fragment of genomic DNA, which contained Pdgfc coding exon 2, to create the targeting construct, in which exon 2 was replaced with an SA-IRES-βgeopA expression cassette. We linearized the targeting construct with NotI, electroporated it into R1 ES cells and selected them with G418 as described previously27. We carried out Southern-blot analysis to screen for the presence of a disrupted Pdgfc gene. We used two independently targeted ES cell clones to generate chimeric mice that subsequently transmitted the mutation through the germ line. The phenotypes of Pdgfc−/− newborns derived from both targeted ES cell lines were indistinguishable.
Genotyping.
We used PCR and Southern-blot analysis for genotyping. We carried out PCR on DNA from ear-punch samples (primer sequences are available on request). We carried out PCR genotyping of Pdgfa mutant embryos as described previously13 and carried out Southern-blot analysis using standard protocols.
Histology, electron microscopy and skeletal staining.
We dissected embryos at different stages and fixed them in 4% paraformaldehyde at 4 °C overnight. We embedded them in paraffin, sectioned them at a thickness of 5 μm and then stained them with hematoxylin and eosin. We carried out transmission electron microscopy and scanning electron microscopy as described28. For skeletal staining, we fixed skinned and eviscerated newborn mice in 95% ethanol for 1–2 d and then stained them with alcian blue and alizarin red as previously described29.
In situ hybridization and immunohistochemistry.
We carried out RNA in situ analysis of whole-mount embryos and frozen tissue sections according to established protocols15. Immunostaining was done as described28.
Derivation of Pdgfc−/− Pdgfa−/− ES cells and tetraploid aggregation.
Pdgfc+/− Pdgfa+/− mice were intercrossed, and the 2.5-d.p.c. blastocysts were flushed and then cultured in ES culture medium as described30. Inner cell mass outgrowths were then disaggregated into small, multicellular clumps that were cultured further until ES cell colonies developed. We picked colonies and cultured them to establish cell lines. We genotyped cell lines for Pdgfc and Pdgfa using PCR. We carried out tetraploid aggregation to obtain entirely ES cell–derived embryos as described17.
Note: Supplementary information is available on the Nature Genetics website.
References
Li, X. et al. PDGF-C is a new protease-activated ligand for the PDGF α-receptor. Nat. Cell Biol. 2, 302–309 (2000).
Gilbertson, D.G. et al. Platelet-derived growth factor C (PDGF-C), a novel growth factor that binds to PDGF α and β receptor. J. Biol. Chem. 276, 27406–27414 (2001).
Taya, Y., O'Kane, S. & Ferguson, M.W. Pathogenesis of cleft palate in TGF-β3 knockout mice. Development 126, 3869–3879 (1999).
Martinez-Alvarez, C. et al. Medial edge epithelial cell fate during palatal fusion. Dev. Biol. 220, 343–357 (2000).
Raich, W.B., Agbunag, C. & Hardin, J. Rapid epithelial-sheet sealing in the Caenorhabditis elegans embryo requires cadherin-dependent filopodial priming. Curr. Biol. 9, 1139–1146 (1999).
Vasioukhin, V., Bauer, C., Yin, M. & Fuchs, E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 100, 209–219 (2000).
Betsholtz, C., Karlsson, L. & Lindahl, P. Developmental roles of platelet-derived growth factors. Bioessays 23, 494–507 (2001).
Hoch, R.V. & Soriano, P. Roles of PDGF in animal development. Development 130, 4769–4784 (2003).
Soriano, P. The PDGF α receptor is required for neural crest cell development and for normal patterning of the somites. Development 124, 2691–2700 (1997).
Tallquist, M.D. & Soriano, P. Cell autonomous requirement for PDGFRα in populations of cranial and cardiac neural crest cells. Development 130, 507–518 (2003).
Fruttiger, M. et al. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development 126, 457–467 (1999).
Karlsson, L., Bondjers, C. & Betsholtz, C. Roles for PDGF-A and sonic hedgehog in development of mesenchymal components of the hair follicle. Development 126, 2611–2621 (1999).
Bostrom, H. et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85, 863–873 (1996).
Karlsson, L., Lindahl, P., Heath, J.K. & Betsholtz, C. Abnormal gastrointestinal development in PDGF-A and PDGFR-α deficient mice implicates a novel mesenchymal structure with putative instructive properties in villus morphogenesis. Development 127, 3457–3466 (2000).
Ding, H. et al. The mouse Pdgfc gene: dynamic expression in embryonic tissues during organogenesis. Mech. Dev. 96, 209–213 (2000).
Aase, K., Abramsson, A., Karlsson, L., Betsholtz, C. & Eriksson, U. Expression analysis of PDGF-C in adult and developing mouse tissues. Mech. Dev. 110, 187–191 (2002).
Nagy, A., Rossant, J., Nagy, R., Abramow-Newerly, W. & Roder, J.C. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. USA 90, 8424–8428 (1993).
Leveen, P. et al. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 8, 1875–1887 (1994).
Soriano, P. Abnormal kidney development and hematological disorders in PDGF β-receptor mutant mice. Genes Dev. 8, 1888–1896 (1994).
Murray, J.C. Gene/environment causes of cleft lip and/or palate. Clin. Genet. 61, 248–256 (2002).
Suzuki, K. et al. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat. Genet. 25, 427–430 (2000).
Kondo, S. et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 32, 285–289 (2002).
van den Boogaard, M.J., Dorland, M., Beemer, F.A. & van Amstel, H.K. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat. Genet. 24, 342–343 (2000).
Proetzel, G. et al. Transforming growth factor-β 3 is required for secondary palate fusion. Nat. Genet. 11, 409–414 (1995).
Kaartinen, V. et al. Abnormal lung development and cleft palate in mice lacking TGF-β 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 11, 415–421 (1995).
Marazita, M.L. et al. Genome scan for loci involved in cleft lip with or without cleft palate, in Chinese multiplex families. Am. J. Hum. Genet. 71, 349–364 (2002).
Gertsenstein, M., Lobe, C. & Nagy, A. ES cell-mediated conditional transgenesis. Methods Mol. Biol. 185, 285–307 (2002).
Ding, H. et al. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 61, 3826–3836 (2001).
Nagy, A. et al. Dissecting the role of N-myc in development using a single targeting vector to generate a series of alleles. Curr. Biol. 8, 661–664 (1998).
Schoonjans, L. et al. Improved generation of germline-competent embryonic stem cell lines from inbred mouse strains. Stem Cells 21, 90–97 (2003).
Acknowledgements
We thank P. Rowe and J. Rossant for reading the manuscript. This work was supported by grants from the National Cancer Institute of Canada Terry Fox Foundation (A.N.); the National Health and Medical Research Council of Australia (P.P.L.T.); and the National Institutes of Health, National Institute of Dental and Craniofacial Research (M.L.M. and L.L.F.). H.D. was supported by a fellowship from the Canadian Institute of Health Research and a developmental grant from the American Muscular Dystrophy Association. H.D. holds a Canada Research Chair. P.P.L.T. is a National Health and Medical Research Council Senior Principal Research Fellow and A.N. is a Senior Scientist of the Canadian Institute of Health Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Table 1
Incidence of fusion of palate shelves isolated from E13.5 pdgf-c mutant embryos in vitro. (PDF 2 kb)
Rights and permissions
About this article
Cite this article
Ding, H., Wu, X., Boström, H. et al. A specific requirement for PDGF-C in palate formation and PDGFR-α signaling. Nat Genet 36, 1111–1116 (2004). https://doi.org/10.1038/ng1415
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1415
This article is cited by
-
Isocitrate dehydrogenase 1 mutation drives leukemogenesis by PDGFRA activation due to insulator disruption in acute myeloid leukemia (AML)
Leukemia (2023)
-
Neuron-to-vessel signaling is a required feature of aberrant stem cell commitment after soft tissue trauma
Bone Research (2022)
-
PDGF receptor mutations in human diseases
Cellular and Molecular Life Sciences (2021)
-
Blocking PDGF-CC signaling ameliorates multiple sclerosis-like neuroinflammation by inhibiting disruption of the blood–brain barrier
Scientific Reports (2020)
-
Transcriptional analysis of cleft palate in TGFβ3 mutant mice
Scientific Reports (2020)
