
Abstract
Telomerase is a ribonucleoprotein complex that is required to synthesize DNA repeats at the ends of each chromosome. The RNA component of this reverse transcriptase is mutated in the bone marrow failure syndrome autosomal dominant dyskeratosis congenita. Here we show that disease anticipation is observed in families with this disease and that this is associated with progressive telomere shortening.
Similar content being viewed by others

Main
In most somatic cells, the ends of each chromosome, the telomeres, shorten with each cell division owing to incomplete replication of the lagging strand of DNA. To compensate for this telomere loss, germ cells and some stem cells activate a reverse transcriptase called telomerase that synthesizes telomeric repeats onto the ends of each chromosome to maintain telomere lengths at an equilibrium1. The possible relationship between telomere lengths, their rate of erosion and age-related disease has evoked considerable interest.
Dyskeratosis congenita is a multisystem disorder characterized by cutaneous abnormalities, bone marrow failure and an increased predisposition to cancer2. Mutations in dyskerin, which is involved in ribosomal RNA processing3 and in the telomerase complex4, are responsible for the X-linked form of this disease5. Families with autosomal dominant inheritance of dyskeratosis congenita (AD-DC) have mutations in the gene encoding the RNA component of telomerase (TERC)6. These mutations seem to give rise to the disease through haploinsufficiency, through the absence of a 3′ end, impaired RNA accumulation or a catalytic defect7.
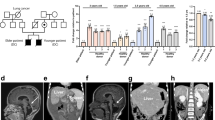
We investigated the telomere lengths and the disease status of 27 affected individuals from eight families with AD-DC (Fig. 1a). In these families, affected individuals are heterozygous with respect to mutations in TERC. Three of the eight families and the mutations they carry were previously described6: family DCR063 (408C→G), family DCR082 (107–108GC→AG) and family DCR101 (3′ deletion, from nucleotide 378 onward). In two of the eight families, the index case presented with aplastic anemia: family DCR172 (72C→G) and family DCR174 (110–113 deletion GACT)8. Of the three mutations described here for the first time, two were detected by denaturing HPLC and direct sequencing of TERC: a CT deletion at nucleotides 96 and 97 (Supplementary Fig. 1 online) and the nucleotide substitution 143G→A (Supplementary Fig. 1 online) in families DCR147 and DCR199, respectively. In family DCR083, a 2,980-bp deletion extends from nucleotide 316 of TERC, 5′ to nucleotide 835 in the 3′ untranslated region of the neighboring gene, which encodes the actin-related protein M1 (Supplementary Fig. 1 and Supplementary Note online). This lesion would abolish TERC expression on one allele in affected individuals. Figure 1b shows the location of the TERC mutations found in the eight families.

(a) Open circles and squares represent normal females and males. Black circles and squares represent affected females and males. Gray circles and squares represent asymptomatic females and males with TERC mutations. The age of disease onset for affected individuals or age of investigation (in brackets) for asymptomatic affected individuals is shown. (b) Model of TERC showing the locations of the mutations found in eight families with AD-DC. (c) Telomere length (measured as a TRF that includes ∼8 kb subtelomeric DNA) is plotted against age for 87 unaffected individuals (open circles) and the 27 individuals with AD-DC (filled circles). A line of best fit is drawn through the normal points. (d) The change in age-adjusted telomere length measurement between parent-child combinations (ΔTELchild − ΔTELparent) in normal and TERC+/− families. Bars show median values. (e) Southern-blot analysis of telomere length. Family trees of families DCR174, DCR199 and DCR147 (from left to right) are drawn above the appropriate lanes. The age of each individual at the time of TRF analysis is shown. Note the bimodal distribution of telomere length in three members of family DCR147, including the unaffected father.
In these eight families, the disease becomes more severe in succeeding generations (Table 1). We have information for 12 affected parents and 15 affected children. Of the affected parents, 7 of 12 are asymptomatic, ranging in age from 36 to 61 y. In these cases, AD-DC was diagnosed only by the identification of a TERC mutation; subtle signs of the disease were often detected subsequently (Table 1). For the five remaining affected parents, the median age at which disease features were first identified was 37 y. Of the affected children, only 5 of 15 remain asymptomatic: they are aged 3, 7, 11, 14 and 20 y and were diagnosed only through mutation analysis. For the remaining ten affected children, symptoms of dyskeratosis congenita presented at a median age of 14.5 y. These data indicate that there is anticipation in the clinical expression of AD-DC, with onset of disease features being, on average, two decades earlier in the children than in their parents.
We next investigated whether the telomere length has a role in the process of disease anticipation. We measured the length of the telomere as a terminal restriction fragment (TRF) by Southern-blot analysis using a subtelomeric probe from the long arm of chromosome 7 (refs. 9,10; Supplementary Methods online). The value obtained was age-adjusted by comparing it with the line of best fit through TRF measurements of 87 normal individuals (Fig. 1c). In this way, we calculated an age-adjusted value of telomere length, called ΔTEL (the difference between the actual value and the predicted value), for each individual11. For all parents and children with TERC mutations, the ΔTEL was a negative value (Table 1), highlighting the fact that their telomeres are significantly shorter than normal (P < 0.0001 for both parents and children versus normal controls, Mann-Whitney). Looking at the difference in telomere length between parents and children (ΔTELchild − ΔTELparent) in 15 transmissions of TERC mutations (Table 1 and Fig. 1d), we found that telomeres were significantly shorter in the second generation of affected families compared with normal families (14 parent-child measurements in normal families; P = 0.036, Mann-Whitney). This decrease in telomere length in successive generations may be responsible for the clinical anticipation in AD-DC.
During the course of this study, we noticed that a number of individuals had a bimodal distribution of telomere length (Fig. 1e). We observed this in 8 of 87(9%) of the normal DNA samples. In the AD-DC families, 6 of 27 (22%) affected individuals (from three of the eight families) and 7 of 13 (54%) children who did not inherit the mutation from an affected parent (from the same three families) had a bimodal pattern of telomere length. A bimodal distribution was previously observed in human fibroblast cell lines where the short and long telomeres are linked to maternal and paternal alleles. The difference in telomere length between the two alleles seems to be maintained from the zygote throughout development12.
The only convincing mechanism of disease anticipation in humans described so far involves a genetic change, namely the expansion of triplet repeats observed in several neurodegenerative disorders13. In the families described here, the genetic lesion has remained the same. We propose that its impact on the inherited telomere length has led to presentation of disease at a younger age in succeeding generations. There are clear analogies here with Terc knockout mice14. Parental mice have very long telomeres, and in the first generation, Terc−/− mice are asymptomatic. Features of telomere shortening, which overlap the clinical features seen in dyskeratosis congenita, develop only in the fourth generation. By the sixth generation these mice become infertile. Heterozygous Terc+/− mice are asymptomatic but have a defect in their ability to elongate telomeres in interspecies crosses15. Our study shows that people who are heterozygous with respect to TERC mutations can remain asymptomatic well into adulthood. But this haploinsufficiency also causes dyskeratosis congenita, the severity of which seems to increase as telomeres shorten through the generations.
All samples used in this study were obtained with informed consent and with the approval of the Research Ethics Committee of the Hammersmith Hospitals NHS Trust.
Note: Supplementary information is available on the Nature Genetics website.
References
Greider, C.W. Ann. Rev. Biochem. 65, 337– 365 (1996).
Dokal, I. Br. J. Haematol. 110, 768– 779 (2000).
Tollervey, D. & Kiss, T. Curr. Opin. Cell Biol. 9, 337– 342 (1997).
Mitchell, J.R., Wood, E. & Collins, K. Nature 402, 551– 555 (1999).
Heiss, N.S. et al. Nat. Genet. 19, 32– 38 (1998).
Vulliamy, T. et al. Nature 413, 432– 435 (2001).
Fu, D. & Collins, K. Mol. Cell 11, 1361– 1372 (2003).
Vulliamy, T., Marrone, A., Dokal, I. & Mason, P.J. Lancet 359, 2168– 2170 (2002).
Brown, W.R. et al. Cell. 63, 119– 132 (1990).
Notaro, R., Cimmino, A., Tabarini, D., Rotoli, B. & Luzzatto, L. Proc. Natl. Acad. Sci. USA 94, 13782– 13785 (1997).
Brummendorf, T., Maciejeweski, J.P., Mak, J., Young, N.S. & Lansdorp P.M. Blood 97, 895– 900 (2001).
Baird, D.M., Rowson, J., Wynford-Thomas, D. & Kipling, D. Nature Genet. 33, 203– 207 (2003).
Lindblad, K. & Schalling, M. Semin. Neurol. 19, 289– 299 (1999).
Blasco, M.A. et al. Cell 91, 25– 34 (1997).
Hathcock, K.S. et al. Proc. Natl. Acad. Sci. USA 99, 3591– 3596 (2002).
Acknowledgements
We thank the clinicians and families for providing samples, N. Killeen for technical assistance and the Wellcome Trust for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Vulliamy, T., Marrone, A., Szydlo, R. et al. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet 36, 447–449 (2004). https://doi.org/10.1038/ng1346
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1346
This article is cited by
-
Genetics of human telomere biology disorders
Nature Reviews Genetics (2023)
-
A case report of dyskeratosis congenita caused by a novel TERC mutation
Annals of Hematology (2023)
-
Genetic Counseling and Family Screening Recommendations in Patients with Telomere Biology Disorders
Current Hematologic Malignancy Reports (2023)
-
Telomere biology disorders
npj Genomic Medicine (2021)
-
The relationship between MUC5B promoter, TERT polymorphisms and telomere lengths with radiographic extent and survival in a Chinese IPF cohort
Scientific Reports (2019)
