
Abstract
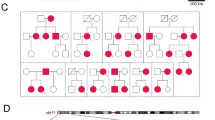
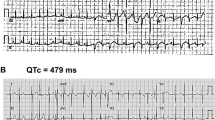
Ion-channel β-subunits are ancillary proteins that co-assemble with α-subunits to modulate the gating kinetics and enhance stability of multimeric channel complexes1,2. Despite their functional importance, dysfunction of potassium-channel p-subunits has not been associated with disease. Recent physiological studies suggest that KCNE1 encodes p-subunits (hminK) that co-assemble with KvLQTI a-subunits to form the slowly activating delayed rectifier K+ (lKs) channel3,4. Because KVLQTI mutations cause arrhythmia susceptibility in the long QT syndrome (LQT)5–7, we hypothesized that mutations in KCNE1 also cause this disorder. Here, we define KCNE1 missense mutations in affected members of two LQT families. Both mutations (S74L, D76N) reduced IKS by shifting the voltage dependence of activation and accelerating channel deactivation. D76N hminK also had a strong dominant-negative effect. The functional consequences of these mutations would be delayed cardiac repolarization and an increased risk of arrhythmia. This is the first description of KCNE1 as an LQT gene and confirms that hminK is an integral protein of the IKS channel.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

References
Rettig, J. et al. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature 369, 289–294 (1994).
Shi, G. et al., β-subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron 16, 843–852 (1996).
Sanguinetti, M.C. et al. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384, 80–83 (1996).
Barhanin, J. et al. KVLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 384, 78–80 (1996).
Wang, Q. et al. Positional cloning of a novel potassium channel gene: KVLQTl mutations cause cardiac arrhythmias. Nature Genet. 12, 17–23 (1996).
Neyroud, N. et al. A novel mutation in the potassium channel gene KVLQT1 causes the JervelI and Lange-Nielsen cardioauditory syndrome. Nature Genet 15, 186–189 (1997).
Splawski, I., Timothy, K.W., Vincent, G.M., Atkinson, D.L. & Keating, M.T. Molecular basis of the long-QT syndrome associated with deafness. N. Engl. J. Med. 336, 1562–1567 (1997).
Keating, M. et al. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science 252, 704–706 (1991).
Jiang, C. et al. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity. Nature Genet. 8, 141–147 (1994).
Swanson, R., Mice, R.E., Folander, K. & Sanguinetti, M.C. The IKS protein, a slowly activating voltage-dependent K+ channel. Semin. Neurosci. 5, 117–124 (1993).
Takumi, T. et al. Alteration of channel activities and gating by mutations of slow ISK potassium channel.. j Biol. Chem. 266, 22192–22198 (1991).
Goldstein, S.A.N. & Miller, C. Site-specific mutations in a minimal voltage dependent K+ channel alter ion selectivity and open-channel block. Neuron 7, 403–408 (1991).
Wang, K.-W. & Goldstein, S.A.N. Subunit composition of minK potassium channels. Neuron 14, 1303–1309 (1995).
Wang, K., Tai, K. & Goldstein, S.A.N. MinK residues line a potassium channel pore. Neuron 16, 571–577 (1996).
Liu, D.-W. & Antzelevitch, C. Characteristics of the delayed rectifier current (lKr and lKs) in canine ventricular epicardial, midmyocardial, and endocardia! myocytes: a weaker lKs contributes to the longer action potential of the M cell. Circ. Res. 76, 351–365 (1995).
Yang, W.P. et al. KvLQTI, a voltage-gated potassium channel responsible for human cardiac arrhythmias. Proc. Natl. Acad. Sci. USA 94, 4017–4021 (1997).
Sanguinetti, M.C., Curran, M.E. & Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG ncodes the lKr potassium channel.. Cell 81, 299–307 (1995).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Splawski, I., Tristani-Firouzi, M., Lehmann, M. et al. Mutations in the hminK gene cause long QT syndrome and suppress lKs function. Nat Genet 17, 338–340 (1997). https://doi.org/10.1038/ng1197-338
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/ng1197-338
This article is cited by
-
Gene mutations in comorbidity of epilepsy and arrhythmia
Journal of Neurology (2023)
-
Clinical utility gene card for: Long-QT syndrome
European Journal of Human Genetics (2021)
-
A cryptic splice-altering KCNQ1 variant in trans with R259L leading to Jervell and Lange-Nielsen syndrome
npj Genomic Medicine (2021)
-
Age-related hearing loss pertaining to potassium ion channels in the cochlea and auditory pathway
Pflügers Archiv - European Journal of Physiology (2021)
-
Exome sequencing in infants with congenital hearing impairment: a population-based cohort study
European Journal of Human Genetics (2020)
