
Abstract
Smith–Magenis syndrome (SMS) is a mental retardation syndrome associated with deletions involving chromosome 17p11.2. Persons with SMS have characteristic behavioral abnormalities, including self-injurious behaviors and sleep disturbance, and distinct craniofacial and skeletal anomalies. We identified dominant frameshift mutations leading to protein truncation in RAI1 in three individuals who have phenotypic features consistent with SMS but do not have 17p11.2 deletions detectable by standard fluorescence in situ hybridization techniques.
Similar content being viewed by others

Main
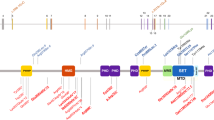
Smith–Magenis syndrome (SMS) is a syndrome of multiple congenital anomalies and mental retardation that encompasses unusual behavioral abnormalities, sleep disturbance, distinct craniofacial and skeletal anomalies and speech delay1,2,3. Most persons with SMS have a large and common deletion of roughly 4 Mb involving chromosome 17p11.2, but many other affected individuals have smaller, atypical deletions involving 17p11.2 (refs. 4–7). Thorough evaluation of affected individuals and their associated deletions has led to several recent studies documenting and refining the SMS critical interval4,6,8. Our most recent evaluation of the critical interval indicates that the minimum deletion region on 17p11.2 associated with the SMS phenotype is about 950 kb and contains roughly 25 genes7. An abbreviated contig containing several known genes in the SMS critical interval is represented in Figure 1a. Although SMS has generally been regarded as a contiguous gene syndrome1,4,6,9, we hypothesized that individuals with features consistent with SMS but in whom no deletion could be detected may have a mutation in a single candidate gene that is primarily responsible for the phenotype.

a, A minimum tiling path of BACs and PACs (solid lines) for the newly refined SMS critical interval7 and cosmids (dotted lines) used for FISH analysis are depicted. Major genes in the SMS critical region are indicated by black circles; RAI1 is indicated by a gray circle. b, The sequences of the deleted and wild-type RAI1 alleles of individual SMS129 are shown, beginning at nt 2,622 of the RAI1 mRNA. The 29-bp deletion, which eliminates a PspOMI recognition site, is highlighted in gray between the solid lines, and four misincorporated amino acids are shown in the deleted allele. The two bands representing the deleted and non-deleted alleles in individual SMS129 were amplified by PCR and sequenced separately. The gel below the pedigree of the family of individual SMS129 shows the exon 3 PCR amplimer (lanes 1, 3, 5, 7) and the digestion of this PCR product with PspOMI (lanes 2, 4, 6, 8). The doublet in the PCR product from individual SMS129 and the uncut, mutated allele evident in the digest are not present in the parents or sister of the affected individual. c, The sequence tracing for the 4929delC deletion and the predicted frameshift on one RAI1 allele in individual SMS156 are shown. A BglI recognition site is abolished by this 4929delC mutation. The results of the exon 4 PCR (lanes 1, 3, 5) and the digestion of this PCR amplimer with BglI (lanes, 2, 4, 6) are represented in the gel shown beneath the family pedigree of individual SMS156. The undigested, mutated allele in individual SMS156 is not present in the parents. d, The sequence tracing for the 1308delC deletion and the predicted frameshift on one RAI1 allele in individual SMS159 is depicted. The parents of individual SMS159 and the 200 control chromosomes analyzed by sequencing of the exon 3 PCR product do not carry this mutation (data not shown). e, The genomic structure for the primary transcript of RAI1 and a major splice variant, as determined by the UCSC genome browser, are shown. Oligonucleotide sequences and specific PCR conditions are available on request.
We identified three such individuals without detectable deletions involving 17p11.2. These individuals were all residents at a facility that has diagnosed over two dozen individuals with SMS and is well known for its work in describing the clinical phenotype. All three individuals were evaluated clinically through the facility's genetics department on multiple occasions (Table 1 and Supplementary Note 1 online). Based on a strong clinical suspicion of SMS, we received samples for further analysis when initial cytogenetics and fluorescence in situ hydridization (FISH) studies for the 17p11.2 deletion were negative. These three individuals, SMS129, SMS156 and SMS159, have many of the typical features of SMS, including craniofacial abnormalities, sleep disturbances and characteristic behaviors, such as onychotillomania (self-mutilation of finger- and toenails), polyembolokoilomania (insertion of foreign objects into body orifices), spasmodic self-hugging and explosive aggressive episodes (Table 1). These individuals were further evaluated for sub-microscopic deletions in 17p11.2 using an extensive series of FISH probes that span the SMS critical region (see Supplementary Note 2 online). Although our FISH analyses do not eliminate the possibility of a small, cryptic deletion in 17p11.2, they do confirm that individuals SMS129, SMS156 and SMS159 do not have deletions of the SMS critical region.
Next we undertook systematic sequencing of genes in individual SMS129 beginning with three genes localized to the SMS critical interval4,6,8: developmentally regulated GTP binding protein 2 (DRG2), RAS dexamethasone-induced 1 (RASD1) and retinoic acid induced 1 (RAI1). We identified no mutations in either RASD1 or DRG2 but found a deletion of 29 bp in exon 3 of RAI1 on one allele in individual SMS129 (Fig. 1b). This deletion was clearly evident as two bands resolved on a 2% agarose gel representing the deleted and non-deleted alleles (Fig. 1b). The deletion produces a frameshift (Fig. 1b) that introduces eight incorrect amino acids followed by a stop codon, truncating the protein. We predict that this deleted allele encodes a truncated and either abnormally functioning or non-functioning RAI1 protein, probably resulting in haploinsufficiency for RAI1.
We then examined individuals SMS156 and SMS159 for mutations in RAI1. In both individuals we identified a deletion on one allele of a single cytosine in a run of Cs (Fig. 1c,d). In individual SMS156, the deleted C occurred in a run of six Cs ending at nucleotide position 4,929 of the RAI1 mRNA. This 4929delC on the coding strand produces a frameshift, introducing 74 incorrect amino acids and truncating the protein (Fig. 1c). Similarly, we found a deletion on one allele of a single cytosine in a run of four Cs ending at nucleotide position 1,308 in exon 3 of the RAI1 (Fig. 1d) in individual SMS159. This deletion also causes a frameshift that incorporates 34 incorrect amino acids beginning at amino-acid position 437, followed by a stop codon and truncation of the protein. None of the parents of these individuals carried any of these mutations (Fig. 1a–c and data not shown), although we cannot rule out mosaicism in the germ cells. We also screened 200 control chromosomes from individuals of European descent and did not detect these mutations in this population (data not shown). The genomic structure of RAI1 and location of the three mutations in the primary transcript are shown in Figure 1e. (See Supplementary Note 3 for the complete RAI1 sequence.)
RAI1 is a novel gene whose cellular role is still unclear. Rai1 was first identified in mouse carcinoma cells when it was upregulated following treatment with retinoic acid10. It is possible that Rai1 is involved in neuronal differentiation, as in situ and immunocytochemistry analysis of adult mouse brain showed that the Rai1 transcript and protein product are localized to neurons10. The RAI1 protein, which contains regions of sequence similarity to the transcriptional coactivator TCF20 (refs. 11,12), may also be localized to the nucleus and stimulate transcription. Our sequence analysis shows that human RAI1 contains a putative bipartite nuclear localization signal beginning at amino-acid positions 1,113 and 1,176 and predicted N-glycosylation sites. As no known DNA-binding motifs have been identified, RAI1 may interact with other DNA-binding proteins to exert its effects on transcription. These processes may be evolutionarily conserved, as the RAI1 protein has approximately 77% overall identity to the mouse Rai1 protein12 and putative RAI1 expressed-sequence tag (EST) homologs have been sequenced from Rattus norvegicus, Bos taurus and Xenopus laevis.
We suggest that SMS may be similar to previously described microdeletion syndromes in which a single gene is implicated in most of the features but other deleted genes may modify the overall phenotype13,14,15. Haploinsufficiency of RAI1 is probably responsible for the behavioral, neurological, otolaryngological and craniofacial aspects of this syndrome, but more variable features such as heart and renal defects are probably due to hemizygosity of other genes in the 17p11.2 region. Finally, we advocate the use of genomic probes that contain RAI1, such as RPCI-1 253P07, for use in FISH to screen individuals who may have SMS. In individuals with strong clinical features and negative FISH analysis, mutation screening may prove informative.
Accession numbers. Human RAI1 mRNA, GenBank AJ271790; human RAI1 protein, GenProt CAC20423; human RAI1 genomic region, GenBank AJ271791 and NT_030843; bc915D19, GenBank AC080148; bc384M20, GenBank AC087163; pc253P07, GenBank AL354000; pc178F10, GenBank AL035367; bc189D22, GenBank AC099988; RAI1 transcript variant KIAA1802, GenBank AB058723. Mouse Rai1 mRNA, GenBank NM_009021; mouse Rai1 protein, NP_033047. Putative RAI1 EST homologs from R. norvegicus, GenBank BU758552; B. taurus, GenBank BE667697; and X. laevis, GenBank BG813716. Human TCF20 mRNA, GenBank XM_040067; mouse Tcf20, GenBank NM_013836.
URL. When putative single-nucleotide polymorphisms (SNPs) were identified, we compared these to known SNPs found in the National Center for Biotechnology Information SNP database (available at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=snp) and SNP information from the genomic contig containing RAI1. Information on Parents and Researchers Interested in Smith–Magenis Syndrome is available at http://www.smithmagenis.org. The GeneReviews online database is available at http://www.genereviews.org. The University of California Santa Cruz genome browser is available at http://genome.ucsc.edu.
Note: Supplementary information is available on the Nature Genetics website.
References
Greenberg, F. et al. Am. J. Hum. Genet. 49, 1207–1218 (1991).
Smith, A.C. et al. Am. J. Hum. Genet. 24, 393–414 (1986).
Greenberg, F. et al. Am. J. Hum. Genet. 62, 247–254 (1996).
Elsea, S.H. et al. Cytogenet. Cell Genet. 79, 276–281 (1997).
Juyal, R.C. et al. Am. J. Hum. Genet. 58, 998–1007 (1996).
Bi, W. et al. Genome Res. 12, 713–728 (2002).
Vlangos, C.N., Slager, R.E., Yim, D.K.C. & Elsea, S.H. Mol. Genet. Metab. (in the press).
Lucas, R.E., Vlangos, C.N., Das, P., Patel, P.I. & Elsea, S.H. Eur. J. Hum. Genet. 9, 892–902 (2001).
Smith, A.C., Dykens, E. & Greenberg, F. Am. J. Med. Genet. 81, 179–185 (1998).
Imai, Y. et al. Brain Res. Mol. Brain Res. 31, 1–9 (1995).
Rekdal, C., Sjottem, E. & Johansen, T. J. Biol. Chem. 275, 40288–40300 (2000).
Seranski, P. et al. Gene 270, 69–76 (2001).
Ewart, A.K. et al. Nat. Genet. 5, 11–16 (1993).
Oda, T. et al. Nat. Genet. 16, 235–242 (1997).
Kishino, T., Lalande, M. & Wagstaff, J. Nat. Genet. 15, 70–73 (1997).
Acknowledgements
We are grateful to L. Schmidt for sharing RAI1 primer sequences; to M. Grotewiel and K. Friderici for critical reading of the manuscript; to T. Jain, L. Park, S. Shunn and D. Lettau for technical assistance; to the Michigan State University Genomics Technology Support Facility and Macromolecular, Sequencing, and Structure Facility for technical support; to the families of individuals with SMS and to Parents and Researchers Interested in Smith–Magenis Syndrome for their persistence and support of our research; to P. Patel for numerous helpful discussions and critical reading of the manuscript; to C.I. Scott, Jr. and E.H. Zackai for initial clinical evaluations; and to A. Smith for discussions regarding the phenotype. This work was funded by a grant from the US National Institutes of Health and with resources from the Michigan State University College of Natural Science Undergraduate Research Support Program and the Howard Hughes Undergraduate Research Support Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Slager, R., Newton, T., Vlangos, C. et al. Mutations in RAI1 associated with Smith–Magenis syndrome. Nat Genet 33, 466–468 (2003). https://doi.org/10.1038/ng1126
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1126
This article is cited by
-
Retinoic acid-induced 1 gene haploinsufficiency alters lipid metabolism and causes autophagy defects in Smith-Magenis syndrome
Cell Death & Disease (2022)
-
Integration of genetic, transcriptomic, and clinical data provides insight into 16p11.2 and 22q11.2 CNV genes
Genome Medicine (2021)
-
Composite Sleep Problems Observed Across Smith–Magenis Syndrome, MBD5-Associated Neurodevelopmental Disorder, Pitt–Hopkins Syndrome, and ASD
Journal of Autism and Developmental Disorders (2021)
-
The behavioural phenotype of Potocki-Lupski syndrome: a cross-syndrome comparison
Journal of Neurodevelopmental Disorders (2018)
-
Comparison of village dog and wolf genomes highlights the role of the neural crest in dog domestication
BMC Biology (2018)
