
Abstract
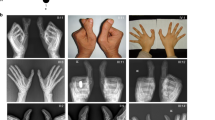
Inherited limb malformations provide a valuable resource for the identification of genes involved in limb development1,2. Brachydactyly type B (BDB), an autosomal dominant disorder, is the most severe of the brachydactylies3 and characterized by terminal deficiency of the fingers and toes. In the typical form of BDB, the thumbs and big toes are spared, sometimes with broadening or partial duplication4,5,6,7,8. The BDB1 locus was previously mapped to chromosome 9q22 within an interval of 7.5 cM (refs 9,10). Here we describe mutations in ROR2, which encodes the orphan receptor tyrosine kinase ROR2 (ref. 11), in three unrelated families with BDB1. We identified distinct heterozygous mutations (2 nonsense, 1 frameshift) within a 7–amino-acid segment of the 943–amino-acid protein, all of which predict truncation of the intracellular portion of the protein immediately after the tyrosine kinase domain. The localized nature of these mutations suggests that they confer a specific gain of function. We obtained further evidence for this by demonstrating that two patients heterozygous for 9q22 deletions including ROR2 do not exhibit BDB. Expression of the mouse orthologue, Ror2, early in limb development indicates that BDB arises as a primary defect of skeletal patterning.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Accession codes
References
Innis, J.W. & Mortlock, D.P. Limb development: molecular dysmorphology is at hand! Clin. Genet. 53, 337– 348 (1998).
Manouvrier-Hanu, S., Holder-Espinasse, M. & Lyonnet, S. Genetics of limb anomalies in humans. Trends Genet. 15, 409–417 ( 1999).
Fitch, N. Classification and identification of inherited brachydactylies. J. Med. Genet. 16, 36–44 (1979).
MacArthur, J.W. & McCullough, E. Apical dystrophy: an inherited defect of hands and feet. Hum. Biol. 4 , 179–207 (1932).
Malloch, J.D. Brachydactyly and symbrachydactyly. Ann. Hum. Genet. 22, 36–37 (1957).
Degenhardt, K.-H. & Geipel, G. Dominant erbliche Perodaktylien in 4 generationen einer Sippe. Z. menschl. Vererb.- u. Konstitutionslehre 32, 227–307 (1954).
Battle, H.I., Walker, N.F. & Thompson, M.W. Mackinder's hereditary brachydactyly: phenotypic, radiological, dermatoglyphic and genetic observations in an Ontario family . Ann. Hum. Genet. 36, 415– 424 (1973).
Houlston, R.S. & Temple, I.K. Characteristic facies in type B brachydactyly. Clin. Dysmorphol. 3, 224– 227 (1994).
Gong, Y. et al. Brachydactyly type B: clinical description, genetic mapping to chromosome 9q, and evidence for a shared ancestral mutation. Am. J. Hum. Genet. 64, 570–577 (1999).
Oldridge, M. et al. Brachydactyly type B: linkage to chromosome 9q22 and evidence for genetic heterogeneity. Am. J. Hum. Genet. 64, 578–585 (1999).
Masiakowski, P. & Carroll, R.D. A novel family of cell surface receptors with tyrosine kinase-like domain. J. Biol. Chem. 267, 26181–26190 (1992).
DeChiara, T.M. et al. Ror2, encoding a receptor-like tyrosine kinase, is required for cartilage and growth plate development. Nature Genet. 24, 271–274 ( 2000).
Oishi, I. et al. Spatio-temporally regulated expression of receptor tyrosine kinases, mRor1, mRor2, during mouse development: implications in development and function of the nervous system. Genes Cells 4, 41 –56 (1999).
Deloukas, P. et al. A physical map of 30,000 human genes. Science 282, 744–746 (1998).
Forrester, W.C., Dell, M., Perens, E. & Garriga, G.A.C. elegans Ror receptor tyrosine kinase regulates cell motility and asymmetric cell division. Nature 400, 881– 885 (1999).
Wilson, C., Goberdhan, D.C.I. & Steller, H. Dror, a potential neurotrophic receptor gene, encodes a Drosophila homolog of the vertebrate Ror family of Trk-related receptor tyrosine kinases. Proc. Natl Acad. Sci. USA 90, 7109–7113 (1993).
Oishi, I. et al. A novel Drosophila receptor tyrosine kinase expressed specifically in the nervous system. J. Biol. Chem. 272, 11916–11923 (1997).
Pawson, T. Protein modules and signalling networks. Nature 373 , 573–580 (1995).
Afzal, A.R. et al. Linkage of recessive Robinow syndrome to a 4 cM interval on chromosome 9q22. Hum. Genet. (in press).
Dib, C. et al. A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380, 152– 154 (1996).
Blair, I.P., Hulme, D., Dawkins, J.L. & Nicholson, G.A. A YAC-based transcript map of human chromosome 9q22.1–q22.3 encompassing the loci for hereditary sensory neuropathy type I and multiple self-healing squamous epithelioma. Genomics 51, 277– 281 (1998).
Wilkie, A.O.M. et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nature Genet. 9, 165–172 (1995).
Oldridge, M. et al. Genotype-phenotype correlation for nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum. Mol. Genet. 6, 137–143 (1997).
Acknowledgements
We thank E. Jowitt, J. Loughlin, H. Santos and K. Temple for their help with earlier stages of this work; S. Butler and N. Elanko for technical assistance; G. Morriss-Kay for discussions; and D. Weatherall for support. The Chromosome Abnormality Database is funded by South East NHSE. This work was funded by Wellcome Trust awards to M.O. and A.O.M.W.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oldridge, M., M Fortuna, A., Maringa, M. et al. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B. Nat Genet 24, 275–278 (2000). https://doi.org/10.1038/73495
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/73495
This article is cited by
-
A novel variant in the ROR2 gene underlying brachydactyly type B: a case report
BMC Pediatrics (2022)
-
Prospects for pharmacological targeting of pseudokinases
Nature Reviews Drug Discovery (2019)
-
A high-throughput integrated microfluidics method enables tyrosine autophosphorylation discovery
Communications Biology (2019)
-
Targeted next-generation sequencing-based molecular diagnosis of congenital hand malformations in Chinese population
Scientific Reports (2018)
-
In-Silico Drug discovery approach targeting receptor tyrosine kinase-like orphan receptor 1 for cancer treatment
Scientific Reports (2017)
