
Abstract
Thellungiella parvula1 is related to Arabidopsis thaliana and is endemic to saline, resource-poor habitats2, making it a model for the evolution of plant adaptation to extreme environments. Here we present the draft genome for this extremophile species. Exclusively by next generation sequencing, we obtained the de novo assembled genome in 1,496 gap-free contigs, closely approximating the estimated genome size of 140 Mb. We anchored these contigs to seven pseudo chromosomes without the use of maps. We show that short reads can be assembled to a near-complete chromosome level for a eukaryotic species lacking prior genetic information. The sequence identifies a number of tandem duplications that, by the nature of the duplicated genes, suggest a possible basis for T. parvula's extremophile lifestyle. Our results provide essential background for developing genomically influenced testable hypotheses for the evolution of environmental stress tolerance.
Similar content being viewed by others

Main
According to phylogenetic studies based on fossil evidence3, the spilt between A. thaliana and the main Brassica group encompassing T. parvula in the subclade Eutremeae is thought to have occurred about 43 million years ago. Both T. parvula and A. thaliana have similar genome sizes, and their close taxonomic relationship provides unique opportunities for tracing evolutionary rearrangements between the two species.
The main goal of this project was to produce a de novo, scaffold-level, gap-free assembly of the T. parvula genome. To achieve this, we used second generation sequencing exclusively, including ROCHE-454 GS FLX Titanium sequencing for its read length advantage and Illumina GA2 sequencing for its higher quality reads. We included varying insert sizes of paired-end libraries in addition to single-end reads (Online Methods). In total, we obtained 7.8 × 109 high quality basepairs, equivalent to ∼50-fold genome coverage. Of these, 85% came from the 454 sequencing (Supplementary Fig. 1 and Supplementary Table 1).
In the absence of genetic maps, with only limited physiological studies4, without prior genome information and with only very limited transcriptome sequences, we used an iterative hybrid approach to construct a draft genome (Online Methods). The result was a total of 1,496 meta-contigs (scaffolds) of merged primary contigs, ranging in size from 1 kb to 13.08 Mb (Table 1). However, unlike typical scaffold sequences, these meta-contigs were free of gaps. Overall, 73% of the length of the T. parvula draft genome was represented in 20 contigs longer than 1.5 Mb, and 85% of the sequenced genome was represented by the largest 60 contigs, each 100 kb or greater in length. Based on flow cytometry of propidium-iodide–stained nuclei5, the T. parvula genome had previously been estimated to be about 160 Mb, or 15% larger than that of A. thaliana. The total size of the curated and assembled T. parvula genome sequence space, however, was 137.09 Mb. This discrepancy is similar to those for A. thaliana (estimated as ∼150 Mb, or 25% longer than the sequenced genome6) and Cucumis sativus (estimated at 30% greater than the draft genome7).
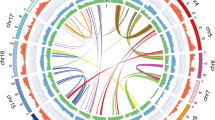
Syntenic regions between T. parvula contigs and other Brassicaceae chromosomes were apparent after aligning T. parvula contigs with the A. thaliana genome (Fig. 1) and chromosome A3 of Brassica rapa8 (Supplementary Fig. 2). The 20 longest contigs covered all five A. thaliana chromosomes, with the exception of positions that approached and included centromeric regions. The largest T. parvula contig, c1 (13.08 Mb), aligned with the entire length of one arm of A. thaliana chromosome 1 (Fig. 1a).

Comparison of the 20 largest T. parvula contigs, c1–c20 (a) and the 40 next largest contigs, c21–c60 (b) with A. thaliana chromosomes. A. thaliana chromosomes 1–5 are depicted as red, green, yellow, purple and blue, respectively, with the centromeric regions indicated by black bands. T. parvula contigs are represented by gray blocks. Regions containing more than 75% similarity over a minimum of 2,000 bp with maximum gap allowance of 1,000 bp are connected with lines of colors matching those used for coloring the A. thaliana chromosomes. Ticks in each chromosome or contig block indicate lengths in 1 Mb. The distributions of protein coding regions and repetitive sequences are shown in the outer circles, with the percentage of protein coding genes, DNA transposons and retrotransposons shown in blue, yellow and orange, respectively, with a window size of 0.1 Mb. In the T. parvula contigs, predicted protein coding genes without BLASTn hits (e value < 0.0001) against the A. thaliana cDNA database are shown in green.
For T. parvula contigs and A. thaliana chromosomes, we annotated repetitive elements (Online Methods). Overall, repetitive sequences amounted to 7.5% of the T. parvula genome based on similarity searches against genomic repeat databases and de novo clustering of repetitive sequences (Supplementary Tables 2,3). Figure 1a and b show repeat distributions in combination with overall sequence alignment comparisons using Circos plots.
Repetitive sequences were distributed unevenly in both species. Repeat-rich sequences were concentrated near the centromeric regions in A. thaliana chromosomes6, as reported for other plant genomes9,10; these sequences were, however, enriched toward the ends of T. parvula contigs (Fig. 1a). As a result of established difficulties in assembling repetitive sequences11, we found repeat-rich sequences more frequently among the smaller T. parvula contigs (Fig. 1b). Thus, the average repeat content in the largest 20 contigs was 5.5%, whereas the next 40 contigs, c21–c60, contained 17.5% repeat content.
We predicted gene models using FGENESH++, GENSCAN and BLAST (see URLs) searches to minimize false positive predictions. We based annotations on sequence similarity identified using independent BLAST searches and the Blast2GO pipeline (Online Methods). We manually inspected predicted open reading frames (ORFs) whose length deviated more than 20% from the putative A. thaliana homologs for exon merging or splitting. T. parvula contained a total of 28,901 predicted protein-coding ORFs. This is about 7% more than A. thaliana, which contains 27,059 protein-coding complementary DNAs (cDNAs) (excluding chloroplast and mitochondrial genes and based on the TAIR9 release). We mapped Illumina short read sequences from the transcriptome of young T. parvula plant tissues to 19,176 of these predicted ORFs (Online Methods and Supplementary Table 4).
The mean size of the predicted ORFs was 1,252 bp, with 71% of the ORFs between 201 bp and 1,500 bp in length (Fig. 2a). This distribution is similar to that of A. thaliana protein-coding cDNAs (Supplementary Fig. 3). The GC contents were substantially higher in exons than in introns and intergenic regions (Table 1). Based on sequence similarity searches to the NCBI nucleotide database, the primary matches for the T. parvula predicted ORFs were most frequently coding regions from Arabidopsis lyrata (53%), A. thaliana (29%) and B. rapa (5%) (Supplementary Table 5). BLASTn searches of T. parvula ORFs against A. thaliana cDNAs identified 25,783 (89%) hits (e value < 0.00001). Among these, 21,523 ORFs were of very similar lengths (80–120%) to their putative A. thaliana homologs (Fig. 2b). The arrangement of predicted ORFs in the T. parvula genome showed extensive macro-synteny with A. thaliana with infrequent rearrangements (Supplementary Table 4), mirroring the genome-wide alignments observed between A. thaliana chromosomes and T. parvula contigs. Each of the 20 largest T. parvula contigs consisted mostly of ORFs that shared sequence similarity with genes from a single A. thaliana chromosome, the exception being contig c3, which shared similarity with genes from three chromosomes (Supplementary Table 6).

(a) Length distribution of predicted T. parvula ORFs. (b) Comparison of T. parvula predicted ORFs with A. thaliana cDNAs showing the highest BLASTn hit score. The ratio of T. parvula ORF length to A. thaliana cDNA length is given as a percentage. In both a and b, the vertical axes and numbers above the bars are counts. Comparison of GO 'biological processes' (c) and GO 'molecular function' categories (d) between A. thaliana cDNAs (At) and T. parvula predicted ORFs (Tp). The GO categories are as defined in TAIR GOslim (see URLs). Categories with significant differences calculated using a χ2 test, as described in the Online Methods, are indicated as *P < 0.05 or **P < 0.01. In c, the GOslim categories 'other metabolic processes' (GO:0008152), 'other physiological processes' (GO:0007582) and 'other biological processes' (GO:0008150) are not shown. The complete list of cDNA and ORF numbers in each of the GO categories and their associated P values are listed in Supplementary Table 8.
A total of 3,118 predicted ORFs had no BLASTn hits to A. thaliana cDNAs even at lowered stringency levels (e value > 0.001). We have listed these as unidentified ORFs in Supplementary Table 4. Notably, these putative ORFs were enriched in regions containing larger numbers of repetitive sequences, possibly indicating T. parvula–specific transposable elements (for example, contigs c17 and c18 in Supplementary Table 6 and the histograms in the outer circle of Fig. 1a). The draft genome also includes 86.6 kb of noncoding RNAs based on sequence searches against microRNA (miRNA) and other noncoding RNA databases (Supplementary Tables 2,7).
We assigned Gene Ontology (GO) terms for the T. parvula predicted ORFs using the Blast2GO pipeline12 and compared them with the A. thaliana transcriptome (Fig. 2 and Supplementary Table 8). In the GO class 'biological processes', subcategories of 'response to abiotic or biotic stimulus' and 'developmental processes' were enriched in T. parvula, whereas genes in the subcategory 'signal transduction' were underrepresented (Fig. 2c). In the GO class 'molecular function', we found the subcategories of 'transporter activity' and 'receptor binding or activity' to be significantly different between the species (Fig. 2d). Among genes annotated as performing transporter activities, the numbers of ATPase and nucleotide, cation and sugar transporters were significantly higher in T. parvula than in A. thaliana (Table 2). These differences may reflect the different habitats and environmental pressures to which the species adapted. ATPase and nucleotide transporters with functions in pH homeostasis and cellular energy generation have, for example, been related to protection under salinity stress13,14, whereas transport and accumulation of soluble sugars or polyols are considered key mechanisms that provide osmotic stress tolerance15. We found the most significant difference in gene copy numbers for transporters of cations other than Na+ and K+, perhaps reflecting the adaptation of T. parvula to soil not only containing saline but also imbalanced in other ions2,16.
Gene copy number variation has also been proposed as a major mechanism of phenotypic differentiation and as reflecting evolutionary adaptation to the environment17,18,19. The T. parvula genome included 1,842 more predicted ORFs than protein coding cDNAs in A. thaliana (Supplementary Fig. 3). Mirroring the observed differences in the GO subcategories, the T. parvula genome contained higher copy numbers of orthologous genes related to stress adaptation, for example, AVP1, HKT1, NHX8 (ref. 20), CBL10 (ref. 21) and MYB47 (Supplementary Table 4).
Gene duplication as a vehicle for evolution has long been hypothesized22, and experimental evidence of this has recently been accumulating7,23,24,25. In both T. parvula and A. thaliana, the major role in generating copy number variation has been played by tandem gene duplication (Fig. 3a) rather than by large gains or losses in segment composition following the A. thaliana and T. parvula divergence after the most recent whole-genome duplication3. We found a total of 1,278 and 1,113 tandem duplication events in the T. parvula and A. thaliana genomes, respectively. Only half of these were shared between the two species (Fig. 3b and Supplementary Table 9). Inspection of the GO class representation of tandem duplications revealed significantly different GO 'biological process' (Fig. 3c) and GO 'molecular function' (Fig. 3d) subcategories. Differences in gene numbers in the subcategories 'response to abiotic or biotic stimulus' and 'developmental processes' (Fig. 2c) were most prominent among genes multiplied by tandem duplication (Fig. 3c), as supported by substantially lower P values (Supplementary Table 8).

(a) Examples of tandem duplications. Examples shown are for the chromosome and contig regions containing HKT1, CBL10 and MYB47. (b) A Venn diagram showing shared and specific tandem duplication events in T. parvula and A. thaliana. We defined a tandem duplication event as the presence of more than one gene with the same annotation in one location or more than one gene in one location separated by not more than one other gene with a different annotation. The numbers of genes involved in the duplication events are given in parentheses. Tandem duplications of genes with the same annotations in both species are counted as shared events. Comparison of the GO 'biological processes' (c) and 'molecular function' categories (d) between T. parvula ORFs and A. thaliana cDNAs for genes showing tandem duplications. The radial axes are the percentages of cDNA or ORFs in each GO category compared to the number of total tandem duplicated cDNA or ORFs. Categories showing significant differences are marked as *P < 0.05 or **P < 0.01. The number of tandem duplicated cDNAs or ORFs in each GO category and P values are listed in Supplementary Table 8. The complete list of tandem duplicated cDNAs and ORFs is presented as Supplementary Table 9.
Finally, Figure 4 and Supplementary Table 10 show the assembly of the T. parvula contigs into the seven chromosomes that characterize this species. The evolution of chromosome structures in Brassicaceae has previously been traced through comparative 'chromosome painting' techniques using BAC-size sequence probes from the A. thaliana genome26. With these techniques, Lysak and colleagues26 identified large genome segments, termed A to X, derived from an ancestral karyotype (n = 8). These ancestral karyotypes can be found in different assemblages in chromosome structures of different Brassicaceae clades26, including A. thaliana27 (Fig. 4a) and Eutremeae28 (n = 7). Using these as guides, the 40 largest T. parvula contigs could be unambiguously assembled into seven chromosomes28 covering 114.39 Mb (83% of the draft genome) (Fig. 4b and Supplementary Table 10). Each of these has a distinct, repeat-rich region, signifying the centromere (Fig. 4c, outer histogram). That the five largest contigs (c1–c5) covered the entire lengths of single chromosome arms attests to the quality of the de novo assembly. It is further noteworthy that the genomic regions in T. parvula contigs c2 and c3, although showing extensive rearrangements compared to the A. thaliana genome sequence, matched distinct ancestral karyotype blocks (Supplementary Fig. 4a, ancestral karyotype blocks R and W for c2, and Supplementary Fig. 4b, ancestral karyotype blocks V, K, L, Q, V′ and X for c3). Thus, our model for the T. parvula chromosomes provides sequence-based evidence for the Lysak model for crucifer species with n = 7, including the clade Eutremeae28. It also defines the boundaries of ancestral karyotype blocks more clearly and suggests more detailed structure than can be captured by chromosome painting experiments alone. This is particularly clear with respect to ancestral karyotype block V, which, based on sequence information, was divided into the blocks V and V′ in the T. parvula genome (Fig. 4b and Supplementary Fig. 4b). Also, ancestral karyotype block I extended to the pericentromeric region of T. parvula chromosome 4 (Fig. 4b,c) rather than falling entirely to one side of the centromere, as previously indicated by the chromosome painting experiments in various crucifer species with n = 7 (ref. 28).

(a) Outline of the ancestral karyotype segments determined by comparative chromosome painting techniques26,27 in A. thaliana chromosomes. The ancestral karyotype segments, denoted A to X, are drawn to scale based on the A. thaliana genome sequence. (b) T. parvula contigs aligned to the Eutremeae (n = 7) karyotype schema26,28 and the ORFs defining the borders of the ancestral karyotype segments. A. thaliana locus IDs showing the highest homology with each ORF are given in parentheses. Shown are T. parvula contigs covering the ancestral karyotype segments. Complete chromosome assignment of the 40 largest contigs, including the contigs covering the centromeric regions, are presented in Supplementary Table 10. (c) Circos plot presenting the assembly of seven chromosomes. The 40 largest T. parvula contigs are shown. The links and histograms in the outer circles showing the distribution of protein coding genes and repetitive sequences were generated as in Figure 1. The ancestral karyotype segments in the A. thaliana chromosomes and T. parvula contigs and the links connecting them are depicted with colors as in a and b.
A number of angiosperm families include extremophile species, although fewer than 10% of all plant species may be classified this way. Extremophiles' presence in evolutionarily distinct lineages reveals genetic complexities that appear to have evolved from the common genetic makeup of all plants. In adaptation to various combinations of environmental stresses, these extremophiles show tolerance of stresses against which crop plants in particular have no defenses. Knowing how extremophiles operate can, however, instruct us about the underlying genetic requisites and mechanisms for successful stress defenses. In this report, we have now shown that it is possible to determine the genome sequence of extremophiles, as well as model glycophytes, exclusively relying on next-generation DNA sequencing tools and de novo assembly.
The availability of the T. parvula genome provides a unique view of chromosome structure, organization and gene complement. Of particular importance is the comparison of this genome with that of the related A. thaliana, which is unquestionably a stress-sensitive species. In our initial analysis, this halophyte, with a genome only ∼15% larger than that of A. thaliana, shows striking differences in gene complement. The differences are partly because of tandem duplications in T. parvula of single copy genes in A. thaliana and preferential amplification of genes with known or assumed functions in stress defense responses. Within these differences, we expect, lie the unique solutions to understanding T. parvula's particular lifestyle and adaptation to its demanding ecological niche. More detailed examination of genome structure, coding complexity, and gene structure and expression in stress response pathways in comparative studies will point the way toward correlating the T. parvula phenotype with its genetic makeup.
URLs.
SeqAnswers online forum, http://seqanswers.com/; GENSCAN, http://genes.mit.edu/GENSCAN.html; BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi; FGENESH++, http://linux1.softberry.com/berry.phtml?topic=fgeneshplus2; TAIR GOslim, ftp://ftp.arabidopsis.org/home/tair/Ontologies/; sff_extract program, http://bioinf.comav.upv.es/sff_extract/; Vmatch suite, http://www.vmatch.de/; AMOS pipeline, http://sourceforge.net/apps/mediawiki/amos/index.php?title=AMOS; Repbase, http://www.girinst.org/repbase/; Plant Repeat database, ftp://ftp.plantbiology.msu.edu/pub/data/TIGR_Plant_Repeats/; TAIR9 cDNA database, ftp://ftp.arabidopsis.org/home/tair/Sequences/blast_datasets/TAIR9_blastsets/TAIR9_cdna_20090619; miRBase, http://www.mirbase.org/; Rfam database, http://rfam.sanger.ac.uk/; GraphPad QuickCalcs, http://www.graphpad.com/quickcalcs/contingency1.cfm.
Methods
Plant material and DNA extraction.
Total DNA was isolated from 10-day-old seedlings of T. parvula. The seeds were derived from a single plant propagated from single seeds over eight successive generations. The original accession was collected from a salt lake in Tuz Golu, central Turkey at an elevation of 905 m above sea level. At the collection site, the soil bulk density was 1.225 g/cm3 with 32.4% salts by weight. Genomic DNA was prepared using the Nucleon Phytopure Genomic DNA Extraction kit (GE Healthcare).
Strategy for a highly contiguous draft genome.
Compared to Sanger sequencing, the shorter reads associated with either 454 or Illumina sequencing manifest decreased connectivity. As a result, considerably deeper coverage is required to generate contiguous assemblies. Deeper coverage alone, however, does not in itself solve the problem of fragmented assemblies; if reads are shorter than a repeat, gaps are unavoidable, and with deeper coverage, accumulated sequencing errors make assembly more computationally challenging. In assembling the T. parvula genome, the problem was mitigated by (i) using reads from different technologies, (ii) using paired reads with different insert lengths to span different repeat lengths and (iii) computationally selecting high quality reads.
Overview of sequencing, assembly and annotation.
Library construction and sequencing were performed in the W.M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign. Random shotgun genomic libraries were constructed according to the manufacturer's recommendations for each of the two pyrosequencing platforms, GS FLX Titanium (454 Life Sciences) and Illumina GA2 (Illumina). Newbler (454-Roche), ABySS29 and minimus2 (ref. 30) were used as the main assembly programs to generate the draft genome, and FGENESH++ (SoftBerry), GENSCAN, BLAST (see URLs) and Blast2GO12 were used to predict and annotate gene models.
DNA library preparation and sequencing.
For 454 pyrosequencing, both shotgun and paired-end libraries were constructed. Genomic DNA was randomly sheared by nebulization to fragments of 500–800 bp in length to construct two shotgun libraries. Additional DNA was processed to construct paired-end libraries with size spans of 3 kb (three libraries), 8 kb (two libraries) and 20 kb (two libraries). All libraries were constructed, clonally amplified and sequenced on the 454 Genome Sequencer FLX-Titanium according to the manufacturer's kits and protocols (454 Life Sciences). Signal processing and base calling were performed using the bundled GS FLX software version 2.0.01.
For Illumina sequencing, genomic DNA was nebulized, and fragments 200–500 bp in length were size selected to construct a shotgun library using the Illumina Genomic DNA Sample Prep kit (Illumina). The library was sequenced on three lanes of a flowcell from one end (single read) for 81 cycles on a Genome Analyzer IIx. The Illumina Pipeline 1.5 was used to generate fastq sequence files from the raw data.
Hybrid genome assembly.
A combined total of 7.8 × 109 bases resulted from sequencing using both platforms. Average read sizes were 355 bp and 80 bp for the 454 and Illumina sequences, respectively. Approximately 85% of all sequences were derived from the 454 sequencing. We followed an iterative approach for assembly starting from raw sequence reads assembled into primary contigs. We used two assembly programs and combined the primary contigs and paired-end data to build scaffolds in successive assemblies. Single and paired-end 454 sequences were assembled using the Roche GS assembler, Newbler (version 2.0.01.14), with a 40 bp minimum overlap and 90% identity. In both instances, reads were first assembled as single-end reads, after which the paired-end information was used to construct scaffolds.
To assemble Illumina reads, we tested both Velvet31 (v1.3) and ABySS29 (v1.2) short-read assemblers using only reads that passed the Illumina chastity filter (base call values for chastity greater than 0.6 in the first 25 cycles); the k-mer size was set to 31 bp, and the coverage cutoff was set to 4. Both assemblers produced comparable results, but ABySS was much faster and was, therefore, chosen for further optimized short read assemblies. We also used Newbler contigs as single reads with the Illumina reads as the input in ABySS. We tested every odd-numbered length from 29 bp to 61 bp as the k-mer size to find the optimal size, meaning that which yielded the longest N50 and the fewest total contigs while maintaining total contig length near the flow-cytrometry–estimated genome size of 160–180 Mb.
Because ABySS can be very sensitive to sequencing errors such as short indels, when using raw 454 reads in ABySS, custom Perl scripts were used to remove any raw 454 read that had homopolymers exceeding 10 bases (all 454 homopolymer error reads cannot be removed but can be minimized). To enable the scaffold generating step in ABySS to proceed when 454 raw paired-ends reads were used, the program sff_extract (see URLs) was used to process the standard flowgram (sff) files generated by the GS FLX sequencer. Different k-mer sizes were selected based on the different paired-end libraries used for scaffolding, but in most instances, the optimum k-mer size found with ABySS was 41.
The collection of contigs and scaffolds created in the primary assemblies was an overlapping set with a high level of redundancy. To select a non-redundant set, we used mkvtree in the Vmatch suite (see URLs) to index the sequences by length; Vmatch was used to cluster sequences, including clusters with size of 1 (singlets). We matched for 100% identity and full coverage of the smaller sequences in pairs. Contigs longer than 1,000 bp were used for further processing. This set was further inspected with all-against-all BLAST searches and the aligner NUCmer in MUMmer 3.22 (ref. 32) to remove duplicate contigs that may have been assembled for the same region of the genome.
The meta-assembly of selected contigs from primary assemblies was carried out with the overlap-layout-consensus assembler, Minimus2 in the AMOS pipeline (see URLs), using a minimum 40-bp overlap with 95% identity. The resulting contigs and singlets were combined and purged of further redundancy, contaminating DNA and mitochondrial and chloroplast DNA using BLAST searches. This resulted in 1,496 contigs with a total length of ∼137 Mb.
Genome annotation.
The T. parvula draft genome was masked for repetitive sequences by RepeatMasker33 searching Repbase 14.01 and with BLASTn using the Plant Repeat database (see URLs). The masked contigs for known repetitive elements were further analyzed with NUCmer and custom scripts to search for long tandem repeats and for T. parvula–specific unclassified, non-exact, long repeats. Any sequences that were found more than five times were considered as repeats in this search.
FGENESH++ (SoftBerry) was used to predict protein coding ORFs in the T. parvula draft genome masked for repetitive sequences, with parameters optimized for dicot plants and protein sequences from the NCBI non-redundant (NR) database as reference. A total of 29,338 ORFs were predicted, of which 437 were further annotated as transposable elements based on BLASTn searches. Genomic regions that contained FGENESH++-predicted ORFs with lengths similar to their Arabidopsis homologs (± 20%) were tested with another gene prediction program, GENESCAN. When the predictions from the two programs deviated for the same genomic region, the ORF closest in length to another known homologous cDNA was taken as the more likely prediction. All genomic contigs and predicted ORFs were searched against NCBI nucleotide and protein databases and TAIR9 cDNA database (see URLs) using BLASTn and BLASTx searches. The predicted proteins were further annotated with the Blast2GO pipeline12 to assign GO and GOslim-plant terms based on NCBI plant databases and InterProScan34. To obtain experimental evidence for our ab initio predictions, we mapped the ORFs to high quality Illumina reads trimmed to 80 bp from a transcriptome sequence library generated from young seedlings. Using the program Bowtie35 with 100% identity to a minimum length of 50 bp and with '-m' set to 1 to ensure unique mapping, we found that 73% of the high quality reads mapped uniquely to the predicted ORFs (Supplementary Table 4). The remaining reads are too repetitive in nature, map to multiple ORFs or contain low complexity regions and are therefore unusable in mapping.
BLAST searches were performed to identify miRNA genes and other RNA genes by searching against the miRBase database of plant miRNA collections (release 16) and the Rfam database (see URLs) (release 10) for other non-coding RNA families including rRNA and tRNA genes.
Statistical analyses.
When comparing the distributions of GO subcategories between A. thaliana and T. parvula (Figs. 2 and 3), two-tailed χ2 tests were used (see URLs). For each GO subcategory, a 2 × 2 contingency table was constructed by recording the numbers of genes included or not included in a subcategory for each species and ranking the statistical significance of the differences.
Accession codes.
The raw reads for this project are deposited in the NCBI SRA project under the accession number SRA026763. The Illumina reads can be accessed under SRX047632 and the 454 reads under SRX032604. The genome assembly is deposited with the NCBI Genome Project ID 63843, and the sequences are deposited with the GenBank ID AFAN00000000.1.
Accession codes
Accessions
GenBank/EMBL/DDBJ
NCBI Reference Sequence
Sequence Read Archive
References
Al-Shehbaz, I.A. & O'Kane, S.L. Placement of Arabidopsis parvula in Thellungiella (Brassicaceae). Novon. 5, 309–310 (1995).
Amtmann, A. Learning from evolution: Thellungiella generates new knowledge on essential and critical components of abiotic stress tolerance in plants. Mol. Plant 2, 3–12 (2009).
Beilstein, M.A. et al. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 107, 18724–18728 (2010).
Orsini, F. et al. A comparative study of salt tolerance parameters in 11 wild relatives of Arabidopsis thaliana. J. Exp. Bot. 61, 3787–3798 (2010).
Oh, D.-H. et al. Genome structures and halophyte-specific gene expression of the extremophile Thellungiella parvula in comparison with Thellungiella salsuginea (Thellungiella halophila) and Arabidopsis. Plant Physiol. 154, 1040–1052 (2010).
Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Huang, S. et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275–1281 (2009).
Mun, J.-H. et al. Sequence and structure of Brassica rapa chromosome A3. Genome Biol. 11, R94 (2010).
Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183 (2010).
International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010).
Alkan, C., Sajjadian, S. & Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65 (2011).
Conesa, A. & Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Intl. J. Plant Genomics 2008, 619832 (2008).
Oh, D.-H. et al. Intracellular consequences of SOS1 deficiency during salt stress. J. Exp. Bot. 61, 1205–1213 (2010).
Gao, F. et al. Cloning of an H+-PPase gene from Thellungiella halophila and its heterologous expression to improve tobacco salt tolerance. J. Exp. Bot. 57, 3259–3270 (2006).
Lugan, R. et al. Metabolome and water homeostasis analysis of Thellungiella salsuginea suggests that dehydration tolerance is a key response to osmotic stress in this halophyte. Plant J. 64, 215–229 (2010).
Inan, G. et al. Salt cress. A halophyte and cryophyte Arabidopsis relative model system and its applicability to molecular genetic analyses of growth and development of extremophiles. Plant Physiol. 135, 1718–1737 (2004).
Sudmant, P.H. et al. Diversity of human copy number variation and multicopy genes. Science 330, 641–646 (2010).
Dassanayake, M. et al. Transcription strength and halophytic lifestyle. Trends Plant Sci. 16, 1–3 (2011).
Hastings, P.J. et al. Mechanisms of change in gene copy number. Nat. Rev. Genet. 10, 551–564 (2009).
Craig Plett, D. & Møller, I.S. Na+ transport in glycophytic plants: what we know and would like to know. Plant Cell Environ. 33, 612–626 (2010).
Quan, R. et al. SCABP8/CBL10, a putative calcium sensor, interacts with the protein kinase SOS2 to protect Arabidopsis shoots from salt stress. Plant Cell 19, 1415–1431 (2007).
Ohno, S. Evolution by Gene Duplication 160 (Springer, New York, New York, USA, 1970).
Hanada, K. et al. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 148, 993–1003 (2008).
Cannon, S.B. et al. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4, 10 (2004).
DeBolt, S. Copy number variation shapes genome diversity in Arabidopsis over immediate family generational scales. Genome Biol. Evol. 2, 441–453 (2010).
Lysak, M.A. & Koch, M.A. Phylogeny, genome, and karyotype evolution of crucifers (Brassicaceae). in Genetics and Genomics of the Brassicaceae (eds. Schmidt, R. & Bancroft, I.). 1–31 (Springer, New York, New York, USA, 2011).
Lysak, M.A. et al. Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc. Natl. Acad. Sci. USA 103, 5224–5229 (2006).
Mandáková, T. & Lysak, M.A. Chromosomal phylogeny and karyotype evolution in x = 7 crucifer species (Brassicaceae). Plant Cell 20, 2559–2570 (2008).
Simpson, J.T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123 (2009).
Sommer, D.D. et al. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 8, 64 (2007).
Zerbino, D.R. & Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Zdobnov, E.M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Acknowledgements
We thank M.P. D'Urzo (Purdue University, West Lafayette, Indiana, USA) for providing plant materials and J.-H. Mun (National Academy of Agricultural Science, Suwon, Korea) for providing the B. rapa chromosome sequence. We also gratefully acknowledge M. Vaughn (University of Texas, Austin, Texas, USA), S. Jackman, M. Krzywinski (Michael Smith Genome Sciences Center, Vancouver, British Columbia, Canada) and SeqAnswers online forum (see URLs) for advice on genome assembly and visualization. Funding has been provided by King Abdullah University of Science and Technology, Thuwal, Saudi Arabia, by the World Class University Program (R32–10148) at Gyeongsang National University, Republic of Korea and the Next-generation BioGreen21 Program (SSAC, PJ008025), Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Contributions
M.D., D.-H.O., H.J.B. and J.M.C. designed, performed, analyzed experiments and wrote the paper; J.S.H. compiled programs and wrote custom scripts; A.H. and S.A. performed sequencing; H.H. prepared materials; D.-J.Y., R.A.B. and J.-K.Z. provided materials and intellectual feedback.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Tables 1, 2, 5 and 6 and Supplementary Figures 1–4. (PDF 948 kb)
Supplementary Table 3
Repetitive sequences in T. parvula draft genome (XLS 4368 kb)
Supplementary Table 4
List of T. parvula predicted ORFs and their annotations (XLS 11262 kb)
Supplementary Table 7
List of non-coding RNAs in T. parvula draft genome (XLS 241 kb)
Supplementary Table 8
List and comparison of GO annotations of the T. parvula predicted ORFs and A. thaliana cDNAs (XLS 5694 kb)
Supplementary Table 9
Tandem local duplications in the T. parvula draft genome and the A. thaliana genome (XLS 1001 kb)
Supplementary Table 10
Assignments of the largest 40 T. parvula contigs in seven chromosomes (XLS 56 kb)
Rights and permissions
About this article
Cite this article
Dassanayake, M., Oh, DH., Haas, J. et al. The genome of the extremophile crucifer Thellungiella parvula. Nat Genet 43, 913–918 (2011). https://doi.org/10.1038/ng.889
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.889
This article is cited by
-
A high-quality chromosome-level Eutrema salsugineum genome, an extremophile plant model
BMC Genomics (2023)
-
Evolutionary diversification of cytokinin-specific glucosyltransferases in angiosperms and enigma of missing cis-zeatin O-glucosyltransferase gene in Brassicaceae
Scientific Reports (2021)
-
Phylogeny and biogeography of the genus Hesperis (Brassicaceae, tribe Hesperideae) inferred from nuclear ribosomal DNA sequence data
Plant Systematics and Evolution (2021)
-
A chromosome-scale reference genome of Lobularia maritima, an ornamental plant with high stress tolerance
Horticulture Research (2020)
-
Arabidopsis HOS15 is a multifunctional protein that negatively regulate ABA-signaling and drought stress
Plant Biotechnology Reports (2020)
