
Abstract
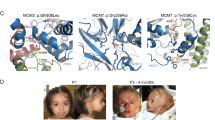
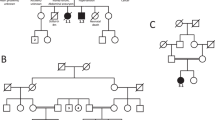
Meier-Gorlin syndrome (ear, patella and short-stature syndrome) is an autosomal recessive primordial dwarfism syndrome characterized by absent or hypoplastic patellae and markedly small ears1,2,3. Both pre- and post-natal growth are impaired in this disorder, and although microcephaly is often evident, intellect is usually normal in this syndrome. We report here that individuals with this disorder show marked locus heterogeneity, and we identify mutations in five separate genes: ORC1, ORC4, ORC6, CDT1 and CDC6. All of these genes encode components of the pre-replication complex, implicating defects in replication licensing as the cause of a genetic syndrome with distinct developmental abnormalities.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Accession codes
References
Bongers, E.M. et al. Meier-Gorlin syndrome: report of eight additional cases and review. Am. J. Med. Genet. 102, 115–124 (2001).
Gorlin, R.J., Cervenka, J., Moller, K., Horrobin, M. & Witkop, C.J. Jr. Malformation syndromes. A selected miscellany. Birth Defects Orig. Artic. Ser. 11, 39–50 (1975).
Meier, Z., Poschiavo & Rothschild, M. Case of arthrogryposis multiplex congenita with mandibulofacial dysostosis (Franceschetti syndrome). Helv. Paediatr. Acta. 14, 213–216 (1959).
Bicknell, L.S. et al. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier Gorlin syndrome. Nat. Genet. advance online publication, doi:10.1038/ng.776 (27 February 2011).
Guernsey, D.L. et al. Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat. Genet. advance online publication, doi:10.1038/ng.777 (27 February 2011).
Neuwald, A.F., Aravind, L., Spouge, J.L. & Koonin, E.V. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43 (1999).
Bowers, J.L., Randell, J.C., Chen, S. & Bell, S.P. ATP hydrolysis by ORC catalyzes reiterative Mcm2–7 assembly at a defined origin of replication. Mol. Cell 16, 967–978 (2004).
Ranjan, A. & Gossen, M. A structural role for ATP in the formation and stability of the human origin recognition complex. Proc. Natl. Acad. Sci. USA 103, 4864–4869 (2006).
Méndez, J. & Stillman, B. Perpetuating the double helix: molecular machines at eukaryotic DNA replication origins. Bioessays 25, 1158–1167 (2003).
Chen, S., de Vries, M.A. & Bell, S.P. Orc6 is required for dynamic recruitment of Cdt1 during repeated Mcm2–7 loading. Genes Dev. 21, 2897–2907 (2007).
Jee, J. et al. Structure and mutagenesis studies of the C-terminal region of licensing factor Cdt1 enable the identification of key residues for binding to replicative helicase Mcm proteins. J. Biol. Chem. 285, 15931–15940 (2010).
Herbig, U., Marlar, C.A. & Fanning, E. The Cdc6 nucleotide-binding site regulates its activity in DNA replication in human cells. Mol. Biol. Cell 10, 2631–2645 (1999).
Liu, J. & Krantz, I.D. Cornelia de Lange syndrome, cohesin, and beyond. Clin. Genet. 76, 303–314 (2009).
Sasaki, T. & Gilbert, D.M. The many faces of the origin recognition complex. Curr. Opin. Cell Biol. 19, 337–343 (2007).
Griffith, E. et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 40, 232–236 (2007).
Al-Dosari, M.S., Shaheen, R., Colak, D. & Alkuraya, F.S. Novel CENPJ mutation causes Seckel syndrome. J. Med. Genet. 47, 411–414 (2010).
O'Driscoll, M., Ruiz-Perez, V.L., Woods, C.G., Jeggo, P.A. & Goodship, J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 33, 497–501 (2003).
Kalay, E. et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat. Genet. 43, 23–26 (2011).
Rauch, A. et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science 319, 816–819 (2008).
Acknowledgements
We thank the subjects and their families for participation in this study and all clinicians for contributing samples not included in this manuscript. Also the late Robert J. Gorlin for contributing individual P3 to the study; E.S. Gray for pathology; M. Ansari, S. McKay, S.D. van der Velde-Visser, T.H. Merkx, S. Devroy and the MRC HGU core sequencing service for advice and technical support; E. Maher and colleagues for cytogenetic advice and assistance; and V. van Heyningen, N. Hastie, D. Fitzpatrick, I. Jackson and J. Blow for discussions and comments. This work was supported by funding from the MRC and Lister Institute of Preventative Medicine. A.P.J. is an MRC Senior Clinical Fellow and Lister Institute Prize fellow.
Author information
Authors and Affiliations
Contributions
L.S.B. performed microsatellite genotyping. L.S.B., S.B. and J.S. performed mutation screening of cases and controls with the help of A.L., E.M.H.F.B., L.H.H., H.v.B. and N.V.A.M.K. M.E.H. performed chromosome breakage analysis. E.M.H.F.B. and A.P.J. clinically characterized the Meier-Gorlin syndrome cases and performed review of phenotypes and sample collection. S.A., J.Y.A.-A., M.B., P.A.J.B., H.v.B., J.D., A.Y.E., M.F., A.F., N.K., N.V.A.M.K., J.M., J.M.O., P.S., A.R., I.K.T., A.T., C.A.W. and M.W. contributed clinical cases and clinical data for the study. A.P.J. and L.S.B. wrote the paper with E.M.H.F.B.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Figure 1 and Supplementary Tables 1 and 2. (PDF 924 kb)
Rights and permissions
About this article
Cite this article
Bicknell, L., Bongers, E., Leitch, A. et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet 43, 356–359 (2011). https://doi.org/10.1038/ng.775
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.775
This article is cited by
-
A CRISPR/Cas9-generated mutation in the zebrafish orthologue of PPP2R3B causes idiopathic scoliosis
Scientific Reports (2023)
-
The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome
European Journal of Human Genetics (2023)
-
Comment on: “The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome” by Nielsen-Dandoroff et al.
European Journal of Human Genetics (2023)
-
High throughput sequencing revealed enhanced cell cycle signaling in SLE patients
Scientific Reports (2023)
-
Chromatin-based DNA replication initiation regulation in eukaryotes
Genome Instability & Disease (2023)
