
Abstract
Plague is a pandemic human invasive disease caused by the bacterial agent Yersinia pestis. We here report a comparison of 17 whole genomes of Y. pestis isolates from global sources. We also screened a global collection of 286 Y. pestis isolates for 933 SNPs using Sequenom MassArray SNP typing. We conducted phylogenetic analyses on this sequence variation dataset, assigned isolates to populations based on maximum parsimony and, from these results, made inferences regarding historical transmission routes. Our phylogenetic analysis suggests that Y. pestis evolved in or near China and spread through multiple radiations to Europe, South America, Africa and Southeast Asia, leading to country-specific lineages that can be traced by lineage-specific SNPs. All 626 current isolates from the United States reflect one radiation, and 82 isolates from Madagascar represent a second radiation. Subsequent local microevolution of Y. pestis is marked by sequential, geographically specific SNPs.
Similar content being viewed by others


Main
Pandemic infectious diseases have accompanied humans since their origins1 and have shaped the form of civilizations2. The designation 'plague' refers to a human invasive disease caused by Y. pestis that is usually fatal without antimicrobial treatment. However, most of the primary hosts for Y. pestis are rodents of various species, in which sylvatic cycles of disease depend on transmission by species-specific flea vectors3. Epidemic expansions of endemic sylvatic disease can result in epidemics and global pandemics of human plague4. Europe was devastated by Justinian's plague (in the years 541–767) and the Black Death (1346 through the eighteenth century)5,6, the latter of which also ravaged China7. Plague resurged in China in 1894, spreading from the Yunnan province to Hong Kong and then via marine shipping to diverse global destinations including India, Europe, Africa and the Americas7. However, direct genetic insights into these historical events were lacking until now.
Y. pestis was subdivided into the biovars Orientalis, Medievalis, Antiqua and Pestoides by their abilities to ferment particular sugars and reduce nitrate8. Genetically, Y. pestis is a monomorphic clone of its more diverse parental species, Yersinia pseudotuberculosis9. Only 76 synonymous SNPs were found in 3,250 orthologous coding sequences from the first three Y. pestis genomes10. These SNPs defined a tree consisting of an ancestral branch 0 and the derived branches 1 and 2. These branches contained populations with distinctive geographic patterns, designated 1.ORI (Orientalis), 2.MED (Medievalis), 1.ANT (African Antiqua) and 2.ANT (east Asian Antiqua). Pestoides (also designated Microtus) were assigned to four other populations, 0.PE1 through 0.PE4. Here we present a global overview of the phylogeographic diversity of Y. pestis and reconstruct historical patterns of plague transmission.
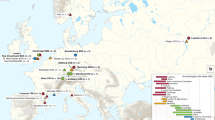
We compared non-repetitive core genomes of 17 isolates of Y. pestis, including 11 that were sequenced for this project (Supplementary Table 1), and found that the phylogenetic patterns of synonymous, non-synonymous and intergenic SNPs were almost identical. The phylogenetic genomes tree in Figure 1 is based on 1,364 SNPs in coding regions that were present in all genomes and was dated using an endemic molecular clock rate based on isolates from Madagascar (Supplementary Table 2). We performed additional mutation discovery in up to 185 kb with 370 isolates from diverse sources and screened 286 isolates for 933 SNPs that were discovered by the combination of genomics and mutation discovery (Supplementary Fig. 1). The resulting SNP assignments were used to calculate a minimal spanning tree (MSTree) and to assign isolates to populations (Fig. 2). The phylogenetic tree and the MSTree share an identical branching order, which is fully parsimonious and reflects unidirectional, clonal evolution from the root to the tips, thus allowing inferences about historical routes of Y. pestis transmission.

Black text, names of genomic sequences (Supplementary Table 1); colored text, branch and population names; gray, ranges of maximal and minimal dates of divergence for individual branches calculated22 with strict mutation rates of 2.9 × 10−9 and 2.3 × 10−8 per site per year (Supplementary Table 2). Comparable results were obtained using intergenic SNPs or a variable clock rate (Supplementary Table 2b). ya, years ago.

Large, bold text, branches 1, 2 and 0; smaller letters, populations (for example, 1.ORI3); lower case letters, nodes (for example, 1.ORI3.a). Strain designations near terminal nodes, genomic sequences. Roman numbers, hypothetical nodes. Gray text on lines between nodes, numbers of SNPs, except that one or two SNPs are indicated by thick and thin black lines, respectively. Six additional isolates in 0.PE1 and 0.PE2b (blue dashes) were tested only for selected, informative SNPs.
A particularly striking aspect of the MSTree is the strong geographical clustering of populations (Supplementary Fig. 2). Isolates from China are scattered in multiple populations over ancestral branch 0, which evolved >2,600 years ago, as well as over branches 1 and 2, which split from branch 0 at least 728 years ago. At the base of branch 0, the Chinese populations 0.PE4 and 0.PE7 are intermingled with the populations 0.PE1 through 0.PE3 from the former Soviet Union (FSU) (Fig. 2). Isolates from outside China or the FSU were only found on branches 1 (1.ORI and 1.ANT) and 2 (2.ANT and 2.MED), suggesting that plague originated in China or the FSU. Consistent with a Chinese source, the average phylogenetic diversity among isolates was greater in China than within other countries (99% bootstrap confidence intervals: China (0.23–0.32), elsewhere (0.029–0.058); Welch t-test: P < 2.2 × 10−16). The genomes tree suggests that 0.PE2 (FSU) is the oldest population, but 0.PE7, isolated in China, is at least as old according to SNP typing and is considerably older according to genomic sequencing (R. Yang, personal communication). Our observations thus suggest that Y. pestis evolved in China and spread to other areas on multiple occasions.
Population 3.ANT, at the end of branch 0 (Fig. 2), is as old as branches 1 and 2 and represents a fourth branch, branch 3, which is apparently restricted to China and Mongolia (R. Yang, personal communication). More recently (>545 years ago), branch 2 split into 2.ANT and 2.MED. This evolutionary separation probably occurred in China with subsequent transmission by land to other areas; all isolates in 2.ANT3 and 2.ANT2 were from China, whereas the terminal nodes in 2.ANT1 were isolated in neighboring Nepal (Fig. 2). Similarly, all isolates in 2.MED3 and 2.MED2 were from China, whereas the terminal nodes in 2.MED1 were from Kurdistan. Isolates in 1.ANT were restricted to east and central Africa, which also requires long-distance travel if Y. pestis evolved in China. The next population on branch 1, 1.IN, consists of three sub-populations (Fig. 2) from western and southern China (Supplementary Figs. 2 and 3).
The youngest population on branch 1, 1.ORI, evolved >210 years ago and spread globally through multiple independent radiations during the third plague pandemic (Supplementary Fig. 3e). The earliest node in 1.ORI gave rise to three sub-branches: 1.ORI1, 1.ORI2 and 1.ORI3. 1.ORI1 reached the United States. 1.ORI2 refers to multiple radiations (radiations iii through ix) that reached Europe, South America, Africa and southeast Asia (Table 1). 1.ORI3 spread to Madagascar and Turkey.
We postulate that sub-branch 1.ORI1 evolved in China because its ancestral node, 1.ORI1.a, contains one isolate from China plus two isolates from Indonesian Java (Supplementary Fig. 4). The next node along the 1.ORI1 sub-branch (1.ORI1.d) contains multiple isolates from northern India, Hawaii and the vicinity of Los Angeles, California, USA. The subsequent evolutionary path in the United States is marked by five strains isolated since 1939 in central California (Supplementary Fig. 4, red and yellow). All 626 other isolates from diverse sources in the western United States belong to descendent nodes derived from the red and yellow nodes. Thus, all extant Y. pestis in the United States seem to be derived from a single import.
We further postulate that sub-branch 1.ORI3 spread from India to Madagascar because its ancestral node, 1.ORI2.a, contains two isolates from India (Supplementary Fig. 5), one of which (strain 195) was isolated in Bombay in the same year (1898) that plague was imported to Madagascar by a plague ship from India11. All 82 strains that were isolated in Madagascar over a period of 80 years fell into two Madagascar-specific clusters radiating from node 1.ORI3.k (blue) or its derived node 1.ORI3.d (red) (Supplementary Fig. 5).
The 1.ORI3.k cluster already existed by 1926, in which year EV76, a widely used attenuated live vaccine strain within the blue cluster, was isolated12. Other members of that cluster were isolated from diverse geographical sources within Madagascar, including the highlands and coastal regions. In contrast, the 1.ORI3.d cluster was restricted to a smaller area in the highlands near Fianarantsoa. Plague first began in this area in 193311, and the oldest member of the 1.ORI3.d cluster is from 1939, suggesting that 1.ORI3.d evolved between 1933 and 1939. Three descendents of 1.ORI3.d were isolated in Turkey.
Clonal microevolution within Y. pestis allows inferences about its evolutionary history, especially when placed in the context of the geographic sources of the isolates and the historical records regarding waves of transmission. One important conclusion is that Y. pestis probably evolved in China. Isolates from China are scattered over all four phylogenetic branches and the average phylogenetic diversity among plague isolates is greater within China than in other countries. Subsequently, Y. pestis has spread from China to other areas on multiple occasions since the origins of branch 0. For example, we infer that Y. pestis on branch 0 spread on multiple occasions to Mongolia, Siberia and central regions of the FSU, which is the most parsimonious explanation for the isolation there of strains with related microsatellite patterns13.
Dates of several branching events predate historical events with which they might be associated for reasons that are explored and analyzed in the Supplementary Note. Genotypes from the Black Death in Europe dating to the mid fourteenth century (∼660 years ago)14 map at or near the split of branches 1, 2 and 3, which occurred >728 years ago. The geographical sources and evolutionary branch order of 2.MED subpopulations, which arose >545 years ago, correspond with points along the former Silk Road15 (Supplementary Fig. 3b), an extensive trade route from China to western Asia between 200 BC and 1400. Other 2.MED1 isolates have been found in western China (R. Yang, personal communication), as well as in Kazakhstan and the Caucasus13, which supports the westward spread of 2.MED from China through trade articles that were carried along the Silk Road.
We also invoke extensive spread of Y. pestis for the 1.ANT1 to 1.ANT3 populations that have only been isolated from east and central Africa. The estimated age of 1.ANT1 (628–6,914 years ago) slightly predates the extensive voyages from China led by Zheng He between 1409 and 1433 (Supplementary Fig. 3a). These voyages involved up to 300 ships, some of which were up to ten times larger than those of contemporary European explorers and carried ∼28,000 crewmen16. It seems highly likely that these ships were infested by rats, which could have transmitted Y. pestis from China to Africa. The geographic locations of 1.ANT isolates are consistent with the terminus of Zheng He's route and suggest progressive evolution during migration from the coast. However, a causal association between 1.ANT in Africa and the voyages by Zheng He remains an unproven hypothesis. Plague may have been introduced to East Africa by an alternative route, such as the limited contacts between East Africa and China facilitated by Arab traders.
The third plague pandemic initially spread from Yunnan to Hong Kong5,6,7 before global dissemination in 1894. This pandemic was caused by Y. pestis isolates from the youngest population on branch 1, 1.ORI, which evolved >210 years ago. As expected, multiple 1.ORI isolates were found in China, including the oldest node of 1.ORI1. Subsequent global dispersion during the third pandemic was associated with multiple independent radiations of the sub-branches 1.ORI1, 1.ORI2 and 1.ORI3.
1.ORI1 spread to northeast India, from which six 1.ORI1.d strains were isolated. These may have been associated with a major plague epidemic in 1899 in Calcutta (now Kolkata), which is thought to have been transmitted from Hong Kong by 1896 (ref. 5). Historical records also document that plague was imported to the United States in 1899 by a plague ship from Hong Kong that docked in Hawaii and then in San Francisco17. Plague broke out soon thereafter in both Hawaii (December, 1899) and San Francisco (March, 1900). Our data pinpoint the origin of modern plague in the continental United States to California, as all extant Y. pestis strains in the United States are the progeny of 1.ORI1.d, which was isolated three times in the vicinity of Los Angeles (Supplementary Fig. 4), where plague-infected squirrels were observed by 1910 (ref. 17). 1.ORI1.d was also isolated in Hawaii, and its ancestor 1.ORI1.a was isolated in China, which is consistent with the historical records. The subsequent evolutionary path in the United States is marked by two descendent nodes containing five strains in central California. All 626 other isolates from diverse sources in the western United States are descendents of those nodes. Thus, all extant Y. pestis in the United States are derived from a single import, possibly corresponding to bacteria introduced to San Francisco in 1899 that then spread to Los Angeles.
1.ORI2.a, a second descendent of 1.ORI1.a, probably also evolved in China because its descendent radiation (radiation ix) spread from the Chinese-Vietnamese border to southern Vietnam and Burma and then back to China (Supplementary Fig. 3e inset). The 1.ORI2.a strain 195 was isolated from Bombay in 1898, which is compatible with historical records showing that Bombay was infected by plague in 1896 by a ship from Hong Kong18. But 1.ORI2.a was also the parent of multiple other radiations that reached Europe, South America, western and southern Africa, and southeast Asia (Table 1 and Supplementary Fig. 3e). For example, radiation viii to Hamburg in Germany and Argentina probably originated in India because plague was imported into Argentina in 1899 from Uruguay by a rice ship from India through Rotterdam19. Several ships carrying plague-infected rats docked in Hamburg soon after 1894 (ref. 20) but did not cause recorded cases of human plague there.
1.ORI2.a also gave rise to sub-branch 1.ORI3 that reached Madagascar. Our data are consistent with one single successful import event from India into Madagascar in 1898, which then differentiated further within Madagascar, finally reaching the highlands in 1921 (ref. 11), where it remains endemic. Alternatively, the original import in 1898 has no extant descendants, and 1.ORI3.k was imported after 1898 but before 1921. Still other scenarios invoking independent imports of the blue and red clusters (Supplementary Fig. 5) after microevolution outside Madagascar are less likely because they would need to account for the restricted geographical specificity of the red cluster within Madagascar. A descendent of 1.ORI3 spread from Madagascar to Turkey because three isolates from Turkey are descendents of nodes that likely evolved within Madagascar. Historical records document two cases of human plague from Madagascar that reached the Middle East in 1931 (ref. 21), which may have been the time period in which transmission to Turkey occurred.
In summary, we present a phylogeny of Y. pestis that covers a large part of its global evolutionary history since an origin in the vicinity of China. We also provide a postulated historical reconstruction for major migrations from east Asia to other continents. The phylogeny of this genetically monomorphic clone is based on an unambiguous reconstruction of the sequential accumulation of approximately 1,000 SNPs that have accumulated in different branches during that phylogenetic history. This extensive SNP-based framework will facilitate future investigations of under-sampled regions, such as Africa and the FSU, for which details are still lacking. It will also help to elucidate the basis of historical pandemics such as Justinian's plague and the Black Death through ancient DNA studies. This study thus provides a basis for more detailed analyses as well as a general paradigm for the reconstruction of historical pandemics.
Methods
Bacterial isolates.
We investigated bacteria from various geographical sources, including 92 isolates from global origins and 98 isolates from China that represent the genetic diversity revealed by biotyping, ribotyping24 and large deletions25, as well as country-specific isolates from Madagascar (82 isolates; Supplementary Table 3) and the United States (651 isolates; Supplementary Table 4). Note, Supplementary Figures 6–10, Supplementary Tables 3–18 and R scripts can be found at http://research.ucc.ie/NG1/index.html. We also tested eight Pestoides isolates10, including the strain Angola, for which no information is available about the source other than its name.
Sources of bacteria for genomic sequencing.
We performed genomic resequencing on 11 Y. pestis isolates (Supplementary Table 1), consisting of four isolates from China (B42003004, E1979001, F1991016 and K1973002), two from Madagascar (MG05-1020 and IP275), one from Uganda (UG05-0454), one from Turkey (IP674), one of ambiguous source (Angola) and two from the United States (CA88, FV-1)26,27,28,29), in order to expand the diversity of genomic polymorphisms beyond that of published genomic sequences30,31,32,33,34,35. Seven of these genomes have been previously reported26,27,28,29. Four others are first reported here, including details of the epidemiological sources and other properties of the bacterial strains.
MG05-1020. Biovar Orientalis, isolated in 2005 in Antananarivo, Madagascar from an 8-year-old male with bubonic plague. MG05-1020 expresses the F1 capsule and is sensitive to Y. pestis–specific bacteriophage, as well as chloramphenicol, trimethoprim and sulfamethoxazole, ciprofloxacin, gentamicin, streptomycin and doxycycline.
IP275 (originally 17/95). Isolated in Madagascar in 1995 from human bubonic plague. IP275 is resistant to eight antibiotics due to a conjugative plasmid36,37.
UG05-0454. Biovar Antiqua. UG05-0454 was isolated in 2004 in Arua, Uganda from a 10-year-old female with bubonic plague. This strain expresses the F1 capsule and is sensitive to Y. pestis–specific bacteriophage, chloramphenicol, trimethoprim and sulfamethoxazole, ciprofloxacin, gentamicin, streptomycin and doxycycline. 3.2 × 103 cfu of this strain kill mice within 4 days.
IP674 (originally T15/1). Biovar Orientalis, isolated in Turkey in 1952.
General genotyping strategy.
In order to avoid phylogenetic discovery bias38, we compared the genomes of 17 Y. pestis isolates (Supplementary Table 1) which represent all known biovars, multiple populations from each of the three known branches and representatives of novel populations from China. Synonymous, non-synonymous and intergenic SNPs were extracted from the genomic comparisons after annotating and excluding potentially repetitive, mobile or hyper-variable regions (Supplementary Table 5).
These comparisons were supplemented with SNP discovery in up to 185 kb by denaturing High Performance Liquid Chromatography (dHPLC)39 with isolates from the different collections. The SNPs discovered within coding regions of the isolates from Madagascar were used to calculate minimal and maximal estimates of a mutational clock rate (Supplementary Fig. 6), which were then used to estimate ranges of dates for the branches in Figure 1 (Supplementary Table 2b).
Finally, 286 isolates were screened by Sequenom MassArray SNP typing for 933 SNPs that had been identified by genomic comparisons and/or SNP discovery, resulting in a minimal spanning tree (MSTree) of clustered nodes (Fig. 2, Supplementary Fig. 7 and Supplementary Table 6). Pestoides isolates were also assigned to this MSTree by typing selected SNPs (Supplementary Table 7). SNP typing identified several homoplastic sites and sequencing errors in the genomic sequences and showed that several isolates represented cross-contamination with vaccine strain EV76 (Supplementary Table 3); these SNPs were all excluded. Thereafter, clustered nodes in the MSTree were assigned to individual populations and subpopulations (Supplementary Table 8 and Supplementary Fig. 7).
Genomic sequencing and assembly.
Isolates IP275, MG05-1020 and UG05-0454 were sequenced at the J. Craig Venter Institute, Rockville, Maryland by random whole genome shotgun sequencing and closure strategies40. Plasmid (pHOS2) and fosmid (pCC1fos) libraries were constructed for each isolate with insert sizes of 4–6 kb and 30–40 kb, respectively. An average of 63,451 high-quality Sanger reads were generated on ABI3730xl as previously described41 and assembled using the Celera assembler42.
IP674 was sequenced at the Wellcome Trust Sanger Institute, using 454 FLX pyrosequencing, and assembled (454/Roche Newbler) into ∼700 contigs (N50 contig size, 13,799 bp) from 784,705 sequence reads of average length 450 bp. Putative SNPs were confirmed by DNA sequencing.
SNP discovery by genomic comparisons.
Chromosomal genomic sequences of Y. pestis (16 sequences) and Y. pseudotuberculosis IP32953 (1 sequence; outgroup) (Supplementary Table 1) were aligned against the well-annotated genome of strain CO9233 using Kodon (Applied Maths) in order to identify non-repetitive SNPs. We used the alignments to identify and exclude all repetitive regions because these can lead to pseudo-SNPs due to faulty alignments or to gene conversion by recombination, resulting in homoplasies43,44. We excluded microsatellites (VNTRs), insertion sequence elements, bacteriophages, homo- and hetero-polymeric repeats, and duplications (the largest of which, DR1-DR2, was 12.1 kb). Additional potential repetitive regions and/or regions that might be under strong diversifying selection were identified by examining 31 bps flanking each potential SNP for three or more polymorphic sites across the 17 Y. pestis genomes. Other repetitive regions were identified by reversed best-hit FASTA searches for duplicated regions containing putative SNPs. These procedures excluded 388 kb (8.3%) from the ∼4.65 Mb CO92 strain genome (Supplementary Table 9).
We also excluded all SNPs that were exclusive to the FV-1 genome because that genome is suspected to contain many sequencing errors, and we also excluded SNPs from strain Angola. Angola contains >708 genome-specific SNPs, which is extraordinarily high for a strain of Y. pestis, and no other isolate was closely related to Angola according to dHPLC. Finally, we excluded SNPs in 1,000 regions spanning ∼600 kb that were lacking in one or more major branches in the tree.
Independent lists of SNPs in non-repetitive regions were also generated with the nucmer module of MUMmer45 from pair-wise alignments to CO92 of 16 Y. pestis genomes (excluding that of FV-1). Differences between the Kodon and MUMmer results were resolved by manual inspection. The remaining SNPs were combined with SNPs detected by dHPLC mutation discovery, resulting in a total of 1,232 biallelic SNPs that were considered suitable for genotyping analyses (Supplementary Fig. 1). For each SNP, the ancestral state was assigned to the nucleotide present within Y. pseudotuberculosis IP32953, and the derived state was assigned to the alternative nucleotide found in Y. pestis.
SNP typing of isolates from the United States.
Thirteen SNPs on branch 1.ORI1 were screened (Supplementary Fig. 4) with Y. pestis DNAs from India (N = 2), Hawaii (N = 2) and diverse sources in western states of continental United States (N = 634) (Supplementary Table 4). SNPs s34 and s59 were screened using TaqMan assays as previously described46. SNPs s1076, s1086 and s1135 were screened using similar, newly designed TaqMan assays. SNPs s691, s729 and s985 were screened using Sequenom MassArray as described above. SNPs s57, s58, s60, s274 and s429 were screened using the melt-MAMA approach47 with newly designed primers (Supplementary Table 10).
SNP typing of Pestoides isolates plus Nich51.
Strains Pestoides A, B, C, D, E and G were tested for 39 SNPs specific for the beginning of branch 0 by the melt-MAMA approach47 (Supplementary Table 10). Those results were combined with published sequencing results10 to provide the SNP calls in Supplementary Table 8. The locations of these isolates in Figure 2 and Supplementary Figure 7 reflect the following conclusions: Pestoides E and G in 0.PE2.b share 12 out of 13 derived SNPs with Pest-F, confirming that 0.PE2.b is closely related to 0.PE2.a (ref. 10), and Pestoides A, B, C and D in population 0.PE1 share six out of ten derived SNPs with 0.PE4 isolates. We also reconfirmed10 that strain Nich51 from the FSU is in 1.ORI by melt-MAMA and PCR tests (Supplementary Table 10), and that it differs from all other known 1.ORI isolates by containing an intact glpD gene.
Cluster assignments.
The merged SNP data were stored as a character set in Bionumerics 5.1 (Applied Maths) and depicted as an MSTree whose branch lengths reflect the numbers of SNP differences between pairs of nodes. However, missing data are interpreted by Bionumerics as equivalent to 0, which leads to artificial nodes and branches due to apparent homoplasies. We therefore assigned each isolate to a node on the basis of unambiguous SNP calls. Where such an assignment was ambiguous due to missing data, the ambiguity was resolved by sequencing (Supplementary Table 11). After unambiguously assigning each isolate to a node, we arbitrarily replaced all remaining missing data for that isolate by the SNP calls that were characteristic of other members of the same node. This strategy is justified because different SNP calls in other parts of the phylogenetic tree would correspond to homoplasies, which are exceedingly rare in Y. pestis.
Homoplasies.
Thirty-six SNPs were considered to represent homoplasies because the same nucleotide change, as confirmed by direct sequencing, was found in at least two independent branches of the MSTree (Supplementary Table 12). Ten additional SNPs were scored as putative homoplasies; in those cases, sequence confirmation was not performed because many isolates had missing Sequenom data.
Branch order of the genomes of Angola and 91001.
Strain Angola evolved before strain 91001 according to four SNPs, but an independent fifth SNP (s595) indicated that 91001 evolved earlier (Supplementary Table 13). On the basis of the former four SNPs, we decided that s595 represents a homoplasy that occurred in strain Angola and excluded it from further analyses.
Accession codes.
Genomic sequences have been deposited under the accession codes listed in Supplementary Table 1. The Sequenom results are available under accession number E-MTAB-213 at EMBL-EBI (http://www.ebi.ac.uk).
Accession codes
References
Linz, B. et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 445, 915–918 (2007).
Diamond, J. Guns, Germs and Steel 1–480 (Jonathan Cape, London, UK, 1997).
Stenseth, N.C. et al. Plague: past, present, and future. PLoS Med. 5, e3 (2008).
Keeling, M.J. & Gilligan, C.A. Metapopulation dynamics of bubonic plague. Nature 407, 903–906 (2000).
Pollitzer, R. Plague studies. 1. A summary of the history and survey of the present distribution of the disease. Bull. World Health Organ. 4, 475–533 (1951).
Devignat, R. Variétés de l′espèce Pasteurella pestis. Nouvelle hypothèse. Bull. World Health Organ. 4, 247–263 (1951).
Wu, L.T. Chapter I: historical aspects. in Plague: A Manual for Medical and Public Health Workers 1–55 (Weishengshu, National Quarantine Service, Shanghai, China, 1936).
Anisimov, A.P., Lindler, L.E. & Pier, G.B. Intraspecific diversity of Yersinia pestis. Clin. Microbiol. Rev. 17, 434–464 (2004).
Achtman, M. Evolution, population structure and phylogeography of genetically monomorphic bacterial pathogens. Annu. Rev. Microbiol. 62, 53–70 (2008).
Achtman, M. et al. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl. Acad. Sci. USA 101, 17837–17842 (2004).
Brygoo, E.R. Epidemiologie de la peste à Madagascar. Archive de l′lnstitut Pasteur de Madagascar 35, 7–147 (1966).
Girard, G. Immunity in plague. Acquisitions supplied by 30 years of work on the “Pasteurella pestis EV” (Girard and Robic) strain. Biol. Med. (Paris) 52, 631–731 (1963).
Li, Y. et al. Genotyping and phylogenetic analysis of Yersinia pestis by MLVA: insights into the worldwide expansion of Central Asia plague foci. PLoS ONE 4, e6000 (2009).
Haensch, S. et al. Distinct clones of Yersinia pestis caused the Black Death. PLoS Pathog 6, e1001134 (2010).
Silkroad Foundation. The Bridge between Eastern and Western Cultures. (2009)〈http://www.silkroadfoundation.org/toc/index.html〉.
Levathes, L. When China Ruled the Seas: The Treasure Fleet of the Dragon Throne, 1405–1433 1–252 (Oxford University Press, New York, 1996).
Link, V.B. A history of plague in United States of America. Public Health Monogr. 26, 1–120 (1955).
Yu, H.L. & Christakos, G. Spatiotemporal modelling and mapping of the bubonic plague epidemic in India. Int. J. Health Geogr. 5, 12 (2006).
Del Rio, A., Zegers, R., Boza, R.D. & Montero, L. Informe sobre la epidemia de peste bubonica. La Chilena di Hijiene IX, 1–7 (1904).
Nocht, B. & Giemsa G. Ueber die Vernichtung von Ratten an Bord von Schiffen als Massregel gegen die Einschleppung der Pest. Arbeiten aus dem Kaiserlichen Gesundheitsamte XX, 6 (1903).
Lauzeral, P. & Millischer, P. A propos de la filiation de deux cas. Bull. Soc. Pathol. Exot. 25, 935–941 (1932).
Drummond, A.J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Adjemian, J.Z., Foley, P., Gage, K.L. & Foley, J.E. Initiation and spread of traveling waves of plague, Yersinia pestis, in the western United States. Am. J. Trop. Med. Hyg. 76, 365–375 (2007).
Guiyoule, A. et al. Recent emergence of new variants of Yersinia pestis in Madagascar. J. Clin. Microbiol. 35, 2826–2833 (1997).
Zhou, D. et al. DNA microarray analysis of genome dynamics in Yersinia pestis: insights into bacterial genome microevolution and niche adaptation. J. Bacteriol. 186, 5138–5146 (2004).
Eppinger, M. et al. Draft genome sequences of Yersinia pestis isolates from natural foci of endemic plague in China. J. Bacteriol. 191, 7628–7629 (2009).
Eppinger, M. et al. Genome sequence of the deep-rooted Yersinia pestis strain Angola reveals new insights into the evolution and pangenome of the plague bacterium. J. Bacteriol. 192, 1685–1699 (2010).
Auerbach, R.K. et al. Yersinia pestis evolution on a small timescale: comparison of whole genome sequences from North America. PLoS ONE 2, e770 (2007).
Touchman, J.W. et al. A North American Yersinia pestis draft genome sequence: SNPs and phylogenetic analysis. PLoS ONE 2, e220 (2007).
Garcia, E. et al. Pestoides F, an atypical Yersinia pestis strain from the former Soviet Union. Adv. Exp. Med. Biol. 603, 17–22 (2007).
Song, Y. et al. Complete genome sequence of Yersinia pestis strain 91001, an isolate avirulent to humans. DNA Res. 11, 179–197 (2004).
Chain, P.S. et al. Complete genome sequence of Yersinia pestis strains Antiqua and Nepal516: evidence of gene reduction in an emerging pathogen. J. Bacteriol. 188, 4453–4463 (2006).
Parkhill, J. et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413, 523–527 (2001).
Deng, W. et al. Genome sequence of Yersinia pestis KIM. J. Bacteriol. 184, 4601–4611 (2002).
Chain, P.S.G. et al. Insights into the genome evolution of Yersinia pestis through whole genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. USA 101, 13826–13831 (2004).
Welch, T.J. et al. Multiple antimicrobial resistance in plague: an emerging public health risk. PLoS ONE 2, e309 (2007).
Galimand, M. et al. Multidrug resistance in Yersinia pestis mediated by a transferable plasmid. N. Engl. J. Med. 337, 677–680 (1997).
Pearson, T., Okinaka, R.T., Foster, J.T. & Keim, P. Phylogenetic understanding of clonal populations in an era of whole genome sequencing. Infect. Genet. Evol. 9, 1010–1019 (2009).
Roumagnac, P. et al. Evolutionary history of Salmonella Typhi. Science 314, 1301–1304 (2006).
Nelson, K.E. et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399, 323–329 (1999).
Eppinger, M. et al. The complete genome sequence of Yersinia pseudotuberculosis IP31758, the causative Agent of Far East scarlet-like fever. PLoS Genet. 3, e142 (2007).
Huson, D.H. et al. Design of a compartmentalized shotgun assembler for the human genome. Bioinformatics 17 (Suppl 1), S132–S139 (2001).
Holt, K.E. et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet. 40, 987–993 (2008).
Lowder, B.V. et al. Recent human-to-poultry host jump, adaptation, and pandemic spread of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 106, 19545–19550 (2009).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004).
Vogler, A.J. et al. Assays for the rapid and specific identification of North American Yersinia pestis and the common laboratory strain CO92. Biotechniques 44, 201 203–204, 207 (2008).
Vogler, A.J. et al. Phylogeography of Francisella tularensis: global expansion of a highly fit clone. J. Bacteriol. 191, 2474–2484 (2009).
Acknowledgements
We gratefully acknowledge technical assistance by R. Nera and A. Doyle and helpful comments from A. Rambaut and D. Falush. Support was provided by grants from the German Army Medical Corps (MSAB15A013) and the Science Foundation of Ireland (05/FE1/B882) to M.A., the National Key Program for Infectious Diseases of China (2008ZX10004-009) and the State Key Development Program for Basic Research of China (2009CB522600) to R.Y. and the US Department of Homeland Security (NBCH2070001; HSHQDC-08-C00158) and US National Institutes of Health (AI065359) to P.K. and D.M.W. Whole genome sequencing of Y. pestis strains IP275, MG05-1020 and UG05-045 was supported by federal funds from the National Institute of Allergy and Infectious Diseases, US National Institutes of Health, Department of Health and Human Services (N01 AI-30071), and sequencing of IP674 was supported by funding for Sanger Institute Pathogen Genomics by the Wellcome Trust. Genomic DNA of Y. pestis MG05-1020 was kindly provided by S. Bearden and M. Schriefer (Centers for Disease Control and Prevention, Fort Collins, Colorado, USA).
Author information
Authors and Affiliations
Contributions
M.A., T.W., D.M.W., P.R., J.R., R.Y. and P.K. designed the study. L.R., J.M.P., R.Y. and E.C. contributed Y. pestis DNA and demographic information. G.M., Y.S., M.E., P.R., M.F., B.K., A.J.V., Y.L., Y.C., P.L. and N.R.T. performed sequencing, SNP discovery, MassArray and SNP testing. G.M., Y.S., C.J.M., M.E., P.R., D.M.W. and P.L. performed bioinformatic analyses of the data. C.J.M., T.J., R.L., F.B. and T.W. performed population genetic analyses. M.A., C.J.M., M.E., P.R., D.M.W., T.J., F.B., P.K., T.W., J.R., R.Y. and E.C. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5, Supplementary Tables 1 and 2 and Supplementary Note (PDF 3326 kb)
Rights and permissions
About this article
Cite this article
Morelli, G., Song, Y., Mazzoni, C. et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat Genet 42, 1140–1143 (2010). https://doi.org/10.1038/ng.705
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.705
This article is cited by
-
Genomic diversity of Yersinia pestis from Yunnan Province, China, implies a potential common ancestor as the source of two plague epidemics
Communications Biology (2023)
-
The source of the Black Death in fourteenth-century central Eurasia
Nature (2022)
-
Subversion of GBP-mediated host defense by E3 ligases acquired during Yersinia pestis evolution
Nature Communications (2022)
-
Molecular characterisation of Mycobacterium avium subsp. paratuberculosis in Australia
BMC Microbiology (2021)
-
Biovar-related differences apparent in the flea foregut colonization phenotype of distinct Yersinia pestis strains do not impact transmission efficiency
Parasites & Vectors (2020)
