
Abstract
Mycobacterium tuberculosis is an obligate human pathogen capable of persisting in individual hosts for decades. We sequenced the genomes of 21 strains representative of the global diversity and six major lineages of the M. tuberculosis complex (MTBC) at 40- to 90-fold coverage using Illumina next-generation DNA sequencing. We constructed a genome-wide phylogeny based on these genome sequences. Comparative analyses of the sequences showed, as expected, that essential genes in MTBC were more evolutionarily conserved than nonessential genes. Notably, however, most of the 491 experimentally confirmed human T cell epitopes showed little sequence variation and had a lower ratio of nonsynonymous to synonymous changes than seen in essential and nonessential genes. We confirmed these findings in an additional data set consisting of 16 antigens in 99 MTBC strains. These findings are consistent with strong purifying selection acting on these epitopes, implying that MTBC might benefit from recognition by human T cells.
Similar content being viewed by others


Main
Infection with M. tuberculosis causes enormous worldwide morbidity and mortality. There were more cases of tuberculosis in 2007 (the last year for which data are available) than at any prior point in world history1. Among the factors that contribute to the continued growth of tuberculosis as a global health problem are the efficiency of human-to-human transmission by the aerosol route, the ability of the causal agent M. tuberculosis to persist and to progress despite development of host immune responses and the absence of a vaccine with reliable efficacy in preventing transmission of the infection. Moreover, although attempts to control tuberculosis through improved identification and treatment of infectious cases have been successful in some settings, similar approaches in other contexts have resulted in increasing rates of resistance to available antituberculosis drugs2. Therefore, new approaches to controlling tuberculosis are essential and would greatly benefit from an improved understanding of the biology of the bacteria and their interactions with their human hosts. In particular, understanding the factors that drive the evolution of M. tuberculosis and allow it to evade host defenses may suggest unique opportunities to develop novel strategies against tuberculosis.
Human tuberculosis is caused by M. tuberculosis and Mycobacterium africanum, which are members of the M. tuberculosis complex (MTBC). In addition to these human-adapted pathogens, MTBC includes various animal-adapted forms, such as Mycobacterium bovis, Mycobacterium microti and Mycobacterium pinnipedii3. To characterize the extent and nature of the forces acting to diversify MTBC, we and others have applied several approaches to phylogenetic analysis of multiple clinical isolates from geographically diverse sources. Using SNPs3,4,5,6 or large sequence polymorphisms7,8,9 as genetic markers has resulted in congruent groupings of human-adapted MTBC into six major lineages and consistent geographical associations for each of these lineages10. In addition, these studies have found strong evidence for a clonal population structure of MTBC, without evidence of ongoing horizontal gene transfer. Analysis of SNPs in a total of 7 megabases of DNA sequence from 89 genes in 108 isolates of MTBC provides strong evidence that MTBC originated in Africa and underwent population expansion and diversification after accompanying ancient human migrations out of Africa, followed by global spread and return to Africa of three particularly successful MTBC lineages through recent waves of travel, trade and conquest3. Together, these studies reveal that MTBC has undergone genetic diversification that corresponds to patterns of human migration, suggesting that distinct lineages have coevolved with distinct human populations7. Moreover, they indicate that further understanding of the mechanisms and consequences of the interactions between MTBC and its human host can be obtained through comparative genomic analyses.
Host-pathogen coevolution is characterized by reciprocal adaptive changes in interacting species11. Host immune pressure and associated parasite immune evasion are key features of this process, often referred to as an 'evolutionary arms race'12,13. Studies in human pathogenic viruses, bacteria and protozoa have revealed that genes encoding antigens tend to be highly variable as a consequence of diversifying selection to evade host immunity14,15,16,17. However, it is unknown whether similar evolutionary mechanisms operate in MTBC and whether the bacteria undergo antigenic variation in response to host immune pressure.
Immunity to tuberculosis in humans, nonhuman primates and mice depends on T lymphocytes18. Among human T lymphocyte subsets, CD4+ T cells are clearly essential for protective immunity to MTBC, as demonstrated by the observation that the incidence of active tuberculosis in people infected with HIV is inversely proportional to the number of circulating CD4+ T cells19. In addition to CD4+ T cell responses, humans infected with MTBC develop antigen-specific CD8+ T cell responses20, and MTBC antigen-specific human CD8+ T cells lyse infected cells and contribute to the killing of intracellular MTBC21. Therefore, there is strong evidence that the adaptive immune system represented by CD4+ and CD8+ T cells is an important mechanism for host recognition and control of MTBC. Recognition of foreign antigens by T lymphocytes depends on binding of short peptide fragments (termed epitopes), derived by proteolysis of foreign proteins, to the major histocompatibility complex (MHC; in humans, termed human leukocyte antigen (HLA)) proteins on the surfaces of macrophages and dendritic cells. CD4+ T cells recognize peptide epitopes bound to MHC class II; CD8+ T cells recognize peptide epitopes bound to MHC class I.
To obtain a better understanding of the effects of human T cell recognition on the diversity of MTBC, and to test the hypothesis that MTBC uses antigenic variation as one mechanism of evading elimination by human immune responses, we determined the genome sequences of 21 phylogeographically diverse strains of MTBC and used those genome sequences to analyze the diversity of 491 experimentally verified human T cell epitopes. This analysis produced the unexpected finding that the known human T cell epitopes are highly conserved relative to the rest of the MTBC genome. These results provide evidence that the relationship between MTBC and its human hosts may differ from a classical evolutionary arms race. The results suggest that developers of new approaches to controlling tuberculosis must take into account the possibility that certain human immune responses may actually benefit MTBC.
Results
A genome-wide phylogeny of human-adapted MTBC
A total of 22 mycobacterial strains were included in this work. To study the sequence diversity of T cell antigens in MTBC, we used Illumina next-generation DNA sequencing to generate nearly complete genome sequences from 20 strains representative of the six main human MTBC lineages, and one strain of Mycobacterium canettii, which is the closest known outgroup of MTBC3,22 (Table 1). In addition, we used the published genome sequence of the H37Rv laboratory strain of M. tuberculosis as a common reference23. For each of the 21 strains newly sequenced, a mean of 6.8 million sequence reads with a mean length of 51 base pairs were generated and mapped to the H37Rv reference genome. On average, the reads covered 98.9% of the 4.4-megabase reference genome (Table 1). The regions not covered primarily included members of the highly GC-rich and repetitive PE/PPE gene families24. A total of 32,745 SNPs were identified, corresponding to an average of one SNP call for every 3 kb of sequence generated. We used a total of 9,037 unique SNPs (each of which occurred in one or several strains) to derive a genome-wide phylogeny of 22 strains (Fig. 1 and Supplementary Fig. 1). Six main lineages could be distinguished with high statistical support. These lineages were completely congruent to the strain groupings previously defined on the basis of genomic deletion analysis and multilocus sequencing3,7,10. The perfect congruence between these different phylogenetic markers further corroborates the highly clonal population structure of MTBC and lack of ongoing horizontal gene transfer in this organism25. Because of the comprehensive nature of genome-scale data, we were able to achieve a higher degree of phylogenetic resolution than in all previous studies. In this new phylogeny, the lineages shown in brown and green in Figure 1 (also known as Mycobacterium africanum) are the most basal groups when compared to the M. canettii outgroup. M. africanum is highly restricted to West Africa for reasons that remain unclear8. However, the fact that the two M. africanum lineages represent the most ancestral forms of human MTBC reinforces the notion that human MTBC originated in Africa3,7.

The tree is rooted with M. canettii, the closest known outgroup. Node support after 1,000 bootstrap replications is indicated. Branches are colored according to the six main phylogeographic lineages of MTBC defined previously3,7,8. Highly congruent topologies were obtained by maximum likelihood and Bayesian inference (Supplementary Fig. 1).
Evolutionary conservation across gene categories
We used these genome sequence data and the phylogeny derived from them to compare the genetic diversity in antigens with that in other experimentally determined gene classes. For comparisons across different gene categories, we divided our data set into three gene sets, including 'essential genes', 'nonessential genes' and 'antigens' (Supplementary Fig. 2 and Supplementary Tables 1 and 2). Antigens were defined by the presence of one or more of 491 experimentally confirmed human T cell epitopes (Supplementary Table 3), which were compiled through the Immune Epitope Database (IEDB) initiative26. The 'essential' gene category was defined on the basis of genome-wide analyses of transposon insertion mutants that were unable to grow on Middlebrook 7H11 agar or in the spleens of intravenously infected mice, as reported previously27,28. We excluded from this analysis genes belonging to the PE/PPE gene family24 and those related to mobile elements, as they are difficult to study using current next-generation DNA sequencing technologies (in total, we excluded 273 (6.8%) of 3,990 genes annotated in the H37Rv reference genome; Supplementary Table 4).
On the basis of evolutionary theory and findings in other bacteria29, we expected that in contrast to nonessential genes, essential genes in MTBC would be under stronger purifying selection and thus would be more evolutionarily conserved. In support of this notion, we observed that, on average, essential genes harbored less nucleotide diversity than nonessential genes (Fig. 2; Mann-Whitney U test P < 0.002). We then compared the rates of synonymous and nonsynonymous SNPs in the essential and nonessential gene categories. The synonymous and nonsynonymous changes were derived by comparison to the most likely recent common ancestor of MTBC, which we inferred on the basis of our new genome-wide phylogeny (Fig. 1 and Supplementary Fig. 1). Because MTBC harbors little sequence diversity, it was necessary to analyze the distribution of synonymous and nonsynonymous SNPs using gene concatenates rather than individual genes. We used two measures of distribution: one based on the number of nonredundant SNPs across all 21 MTBC strains (measure A; Fig. 3 and Table 2 show the ratio of the rates of synonymous and nonsynonymous substitutions (dN/dS) from measure A) and one based on individual pairwise comparisons between each strain and the inferred most likely recent common ancestor (measure B; Table 2 shows dN/dS from measure B). From these analyses, we found that the dN/dS values for essential genes were significantly lower than those for nonessential genes (measure A in Fig. 3 and measure B in Table 2; Mann-Whitney U test P < 0.0001). Together, these data show that in MTBC, essential genes are more evolutionarily conserved than nonessential genes.

Box plot indicates median (horizontal line), interquartile range (box) and minimum and maximum values (whiskers).

We calculated overall dN/dS on the basis of the number of nonredundant synonymous and nonsynonymous changes after comparing each of the 21 MTBC genomes to the inferred most likely recent common ancestor of MTBC. This shows that essential genes are more conserved than nonessential genes and that antigens are as conserved as essential genes. Figures for the epitope and non-epitope regions refer to the calculations after we excluded the three outlier antigens esxH, pstS1 and Rv1986.
Because MTBC interacts with humans through antigen-specific CD4+ or CD8+ T-cells, we would expect T cell antigens to be among the most diverse genes in the genome. Particularly when invoking a coevolutionary arms race and associated immune evasion, we would anticipate that these antigens are under diversifying selection and are more variable than other genes, enabling them to escape T cell recognition. However, when we analyzed the nucleotide diversity in 78 experimentally confirmed human T cell antigens (Supplementary Table 2), we found that they were, on average, not more diverse than essential genes (Fig. 2; Mann-Whitney U test P = 0.12). Moreover, we found that the dN/dS values in these antigens also resembled those of essential genes (measure A in Fig. 3 and measure B in Table 2; Mann-Whitney U test P = 0.77). Thus, human T cell antigens in MTBC do not appear to be under diversifying selection. Instead, purifying selection appears to be the driving selection pressure on these genes.
T cell epitopes are hyperconserved
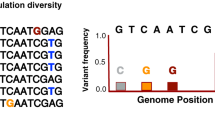
T cell antigens consist of epitope regions that interact with human T cells and non-epitope regions that are not targets of T cell recognition. Hence, we decided to study these regions separately. To this end, we generated a separate concatenate of the epitope regions and another concatenate of all corresponding non-epitope regions. Because little data are currently available in the IEDB with respect to whether these 491 epitopes are recognized by CD4+ or CD8+ T cells, we analyzed them as one class. If immune escape were driving antigen evolution to evade T cell recognition in MTBC, we would expect nonsynonymous changes to accumulate in epitope regions, leading to a high dN/dS. Contrary to this expectation, however, the overall dN/dS of the epitope regions was 0.53, which was similar to that of essential genes and lower than that of nonessential genes (Fig. 3 and Table 2). Moreover, when we analyzed the distribution of amino acid replacements in individual epitopes, we found that a large majority (95%) of the 491 epitopes showed no amino acid change (Fig. 4). Only five epitopes, contained in esxH, pstS1, and Rv1986, harbored more than one variable position (Supplementary Table 5). The higher number of amino acid substitutions in these five epitopes may reflect ongoing immune evasion, but further investigation is needed to determine whether the observed changes are due to immune pressure, other selection pressure(s) or mere random genetic drift3. Because these five epitopes were clear outliers compared to the large majority of T cell epitopes analyzed here, we repeated our dN/dS analysis after excluding the three antigens harboring the five outlier epitopes. Our analysis revealed that the epitope regions had the lowest dN/dS of all gene categories (Fig. 3 and Table 2). Furthermore, when we compared the proportion of nonredundant, nonsynonymous changes in epitope and non-epitope regions, we found that epitopes were less likely than non-epitopes to harbor changes at nonsynonymous sites (measure A in Table 2; χ2, P < 0.05), whereas no difference was observed at synonymous sites (Table 2; χ2, P = 0.89).

This demonstrates the remarkable lack of genetic variability among the regions of the genome that interact with the human immune system.
To further corroborate our finding of hyperconservation of human T cell epitopes in MTBC, we repeated our analysis using a data set from a previous study in which 89 individual genes were sequenced in 99 human-adapted strains representative of the six major global lineages of MTBC3. Sixteen of these 89 genes belonged to the set of T cell antigens analyzed here, including two of the three outlier antigens, esxH and pstS1 (ref. 3). Analysis of this additional data set of 16 antigens in 99 MTBC strains revealed that the epitope regions had an overall dN/dS of 0.74. However, when we excluded the two outlier antigens, the dN/dS dropped to 0.46, which was again lower than the genome-based dN/dS values for essential and nonessential genes (Fig. 3).
Together, our findings strongly suggest that a large proportion of the MTBC genome known to interact with human T cells is highly conserved and is under purifying selection as strong as, or perhaps even stronger than, that of essential genes.
Discussion
In this study of 22 MTBC genomes, we demonstrate that, as expected, essential genes are more conserved than nonessential genes. These results are in agreement with a previous study that analyzed a single genome30. To our surprise, however, we found that a large majority of the currently known T cell antigens are as conserved as essential genes. Furthermore, the epitope regions of these antigen genes are the most highly conserved regions we studied. This observation—that the regions of the genome that interact with the human adaptive immune system appear to be under even stronger purifying selection than essential genes—is inconsistent with a classical model of an evolutionary arms race.
It is possible that the known human T cell epitopes that we found to be hyperconserved represent a select subset of all of the human T cell epitopes encoded in the genome and that certain approaches to epitope identification have favored discovery of hyperconserved epitopes in MTBC. For example, as most, if not all, of the epitope discovery efforts to date have used proteins or peptide sequences of strains from one lineage (lineage 4) and T cells from humans who are likely to have been infected by strains of other lineages, the assays used may have been especially suited to identification of hyperconserved and cross-reactive epitopes. Although further investigation using alternative approaches to epitope discovery may reveal that variable epitopes showing evidence of positive selection exist in the MTBC, it is likely that the large number of epitopes we examined will remain a substantial subset of the total and that future vaccine-development efforts will need to account for the possibility that immune recognition of certain epitopes may actually provide a net benefit to the bacteria.
Lack of antigenic variation and immune evasion has been reported for a number of other human pathogens, including RNA viruses such as measles, mumps, rubella and influenza type C (ref. 31). Theoretical studies have suggested that the absence of immune escape variants in these viruses might be due to structural constraints in viral proteins or negative mutational effects leading to reduced infectivity or transmission31. We cannot exclude the possibility that structural and functional constraints that are independent of T cell recognition contribute to hyperconservation of the regions encoding MTBC peptides recognized by human T cells; however, one important characteristic of the aforementioned viral pathogens is that they spread among young and immunologically naive hosts, which might eliminate the need for immune evasion31. Moreover, infection by these viruses usually results in acute disease followed by elimination of the infection through adaptive immunity, resulting in acquisition of lifelong immunity against reinfection. This further indicates that these viruses are specialized pathogens of immunologically naive hosts. By contrast, MTBC causes chronic and often lifelong infections, and adaptive immunity is usually unable to completely clear the infection18. Furthermore, tuberculosis patients are prone to re-infection32, and mixed infections are also increasingly recognized33. These observations suggest that the biological basis for the lack of antigenic variation in MTBC reported here differs from that proposed for antigenically homogeneous RNA viruses31. In addition, we determined that the fraction of hyperconserved T cell epitopes of the MTBC that are derived from essential genes is indistinguishable from the frequency of essential genes in the MTBC genome as a whole (18% versus 21%, respectively; χ2 = 0.28, P = 0.59), indicating that our results were not skewed by over-representation of T cell epitopes in essential genes. Moreover, the T cell epitopes that we analyzed are present in genes from diverse gene ontologies, and the representation of five main gene categories (defined according to the NCBI Categories of Orthologous Groups (COGs)) was no different in the T cell antigens than in the genome overall (χ2 with 4 degrees of freedom was 5.8, P = 0.21; Supplementary Table 6). Hence, the only identifiable common property of these regions is their recognition by human T lymphocytes. These findings suggest that T lymphocyte recognition is an important factor in hyperconservation of these sequences and that other structural or functional constraints are unlikely to fully account for the lack of sequence variation in these domains.
Our data suggest that T cell epitopes in MTBC are under strong selection pressure to be maintained, perhaps because the immune response they elicit in humans, which is essential for the survival of an individual host, might be partially beneficial to the pathogen. One potential mechanism by which MTBC could benefit from human T cell recognition is that human T cell responses are essential for MTBC to establish latent infection. This notion is supported by the fact that CD4+ T cell–deficient HIV-positive individuals progress rapidly to active disease after infection rather than sustaining latent tuberculosis for prolonged periods34. Latent infection mediated by host T cell responses, with subsequent reactivation to diseaseoften occurring decades after initial infection, is a key characteristic of human tuberculosis and might have evolved as a way for MTBC to transmit itself to later generations of susceptible hosts35. In addition, there is evidence that T cell responses may contribute directly to human-to-human transmission of MTBC. In particular, cavitary tuberculosis, which generates secondary cases more efficiently than other disease forms36, rarely occurs in CD4+ T cell–deficient HIV-positive individuals, and the frequency of cavitary lung lesions in HIV-infected patients with tuberculosis is directly correlated with the number of peripheral CD4+ T cells37. Although the mechanisms of lung cavitation in tuberculosis are poorly understood, these observations suggest that CD4+ T cells directly or indirectly mediate tissue damage in tuberculosis. Together with our finding of epitope hyperconservation, they indicate that certain T cell responses may be detrimental to the host and beneficial to the pathogen. Hence, our findings suggest that MTBC takes advantage of host adaptive immunity to increase its likelihood of spread and that the benefits of enhanced transmission exceed the costs of within-host cellular immune responses to these epitopes. In this way, MTBC may resemble HIV, for which there is evidence that virulence has evolved not to maximize replication of the virus within individual hosts but to maximize the likelihood of its transmission38. Additional studies in humans will be needed to determine whether T cell responses to other epitopes, or specific T cell subsets (for example, Th17 versus Th1) that benefit the host and not the bacteria, can be identified.
One limitation of this study was the exclusion of PE/PPE genes for technical reasons. Some of these genes are known to vary and to encode cell surface–exposed proteins, which has led to the hypothesis that they might be involved in antigenic variation24. However, no direct evidence for this has yet been presented. Future work will need to clarify the function and evolution of PE/PPE genes. By contrast, all the T cell antigens included in this study have been experimentally confirmed26. Furthermore, some of them are being targeted by new tuberculosis diagnostics and vaccines39. Our findings thus have important implications for the development of these new tools. On one hand, the fact that MTBC harbors little sequence diversity in T cell antigens will facilitate the development of diagnostics that are universally applicable across geographical regions where MTBC strains differ8. On the other hand, the possibility that the immune responses induced by vaccine antigens might partially benefit the pathogen suggests that current efforts in vaccine research should be broadened. Most disturbing is the suggestion that vaccine-induced immunity against these conserved epitopes may perversely increase transmission. In light of this, it is noteworthy that the currently available tuberculosis vaccine Bacille-Calmette-Guerin (BCG), which is a live vaccine based on an attenuated from of M. bovis, offers no protection against pulmonary tuberculosis in adults40. Moreover, some clinical trials of BCG have even reported an increased risk of tuberculosis in vaccines compared to unvaccinated individuals41. Thus, in contrast to standard reverse vaccinology, in which the least variable antigens of a genome are targeted42, research into new tuberculosis vaccines should explore more variable regions of the MTBC genome.
Whereas most of the T cell epitopes analyzed here were highly conserved, five epitopes in three antigens harbored a larger number of amino acid changes. The fact that the dN/dS value dropped sharply after we excluded these outlier antigens from the analysis further supports the notion that they are indeed outliers compared to the other antigens. One of the outlier antigens, esxH (Rv0288, also known as TB10.4) is a member of a gene family known to encode a type VII secretion system43. Notably, this antigen is being considered for use in developing a new vaccine against tuberculosis39. Our finding that this particular vaccine antigen harbors a comparatively high number of amino acid substitutions across a panel of global MTBC isolates, even though most of the other vaccine antigens analyzed here are conserved, suggests that strain diversity should be considered during further development of the new vaccine candidates containing esxH (ref. 8).
We detected statistically significant (P < 0.0001) differences in dN/dS between essential, nonessential and antigenic genes. Nevertheless, the individual dN/dS values are high compared to most other bacteria44. Such a high dN/dS was reported previously for MTBC and has been linked to reduced selective constraint against slightly deleterious mutations3. It was proposed that the serial transmission bottlenecks associated with patient-to-patient transmission of MTBC could lead to an increase in random genetic drift. Our new data show that even though the strength of purifying selection in MTBC might be reduced overall compared to other bacteria, it is still acting on, and capable of differentiating between, gene categories.
In summary, we show that T cell epitopes of MTBC are highly conserved and do not reflect any ongoing evolutionary arms race or immune evasion. Instead, the patterns observed might indicate that this highly successful pathogen has developed a distinct evolutionary strategy of immune subversion. Other intracellular bacteria such as Salmonella enterica serovar Typhi show a similar lack of antigenic variation45, suggesting that comparable mechanisms might exist in other pathogens with a similar lifestyle.
Methods
Bacterial strains and DNA sequencing.
Bacterial strains were selected from the six main MTBC lineages defined previously3,7,8 and cultured from single colonies. Genomic DNA was extracted using a standard kit (Qiagen), and 2 μg DNA was used for sequencing with the Illumina Genome Analyzer. Sequencing libraries were constructed using standard kits from Illumina according to the manufacturer's instructions. Libraries for each strain were loaded into a single lane of a flow cell. Sybr green assays were used to test flow cells for optimal cluster density. Single-read sequencing was performed with read lengths of 51 bases and a target coverage of at least 3 million high-quality bases.
SNP detection, filtering of raw SNPs calls and genome assembly.
The program MAQ46 was used to map the Illumina reads of each strain against the reference genome sequence of H37Rv (GenBank NC_000962). The positions with nucleotides differing between the query strain and the reference were recorded as SNPs. Raw SNP calls were filtered to remove low depth coverage and hits with multiple alignment associated with repetitive DNA content. SNPs with depth coverage less than 5 or base quality less than 30 were removed. These parameters were chosen on the basis of previous work carried out in S. enterica serovar Typhi45. SNP lists for individual strains were combined in a single nonredundant data set, and the corresponding base call was recovered for each strain. From the 10,096 initial nonredundant SNPs, we excluded 151 heterozygous base calls and kept 9,945 homozygous SNPs for further analysis. Of these, 8,771 fell into coding regions and 1,174 into noncoding regions.
The orthologs of the 3,990 annotated genes of the H37Rv reference genome were retrieved for each strain using the genome assembly produced by MAQ. Strain-specific gene deletions were identified by the presence of uncalled bases. Multiple gene alignments were generated and used for the different gene sets.
Definition of gene sets.
PE/PPE genes24 or genes described as integrase, transposase or phage related were excluded from the analysis. After excluding 273 of 3,990 genes, we kept 3,717 (93%) for analysis (Supplementary Fig. 2). The list of excluded genes is provided in Supplementary Table 4. The reason we excluded the PE/PPE genes was because current next-generation sequencing technologies generate short sequencing reads that cannot be accurately mapped onto highly repetitive genome regions. We generated a nonredundant data set containing the following three gene categories: essential genes, nonessential genes and antigens. To define antigens, we obtained a list of experimentally confirmed human epitopes from the IEDB database, which we accessed on 26 May 2009 (Immune Epitope Database and Analysis Resource)26. The search criteria were human T cell epitopes described either in M. tuberculosis or M. tuberculosis H37Rv. A total of 582 epitopes were initially identified. We assigned each epitope to an H37Rv gene after inspecting the corresponding bibliographic reference and individual FASTA searches. From the initial 85 antigen genes identified, four were excluded by the exclusion criteria mentioned above. In addition, we excluded one epitope that was defined for Trypanosoma cruzi and referred to M. tuberculosis but which we were unable to identify in the H37Rv genome. Finally, we excluded one antigen with no homolog in H37Rv and another that was present in only two strains. The final number of antigens analyzed was 78 (Supplementary Table 2), including 491 epitope sequences (Supplementary Table 3). As only a minority of the M. tuberculosis epitopes listed in the IEDB have been unambiguously determined to be recognized by CD4+ or CD8+ T cells, we analyzed all human T cell epitopes as one group.
After defining these 78 antigens, we classified the remaining 3,639 genes into essential and nonessential as follows. Essential genes in M. tuberculosis have been defined both for growth in vitro and for growth in vivo27,28. Both experiments used transposon mutagenesis to generate single gene knockouts, followed by transposon site hybridization after growth on 7H11 agar or in mice. On the basis of these studies, we placed a total of 760 genes in the category of essential genes (Supplementary Table 1). The remaining 2,879 genes in the genome were pooled into the non–essential-gene category (Supplementary Table 1 and Supplementary Fig. 2).
Phylogenetic analysis and ancestral reconstruction.
Our phylogenetic analysis was based on all filtered SNPs detected when we compared each strain against the reference H37Rv. Both coding and noncoding SNPs were included. The SNPs were used to infer the phylogenetic relationships between strains using neighbor joining (Fig. 1), maximum likelihood (Supplementary Fig. 1a) and Bayesian (Supplementary Fig. 1b) methodologies. Because some SNP calls were missing from individual strains, we used an ungap alignment for the phylogenetic analysis based on 9,037 positions. The neighbor-joining tree was obtained using MEGA47 with observed number of substitutions as a measure of genetic distance. The maximum-likelihood topology was obtained using PhymL48 with 1,000 bootstrap pseudoreplicates for clade support. We used the Akaike information criterion as implemented by Modeltest49 to select the best-fit model of nucleotide substitution. The Bayesian-inference tree was obtained using two runs of four chains during 1.5 million generations and the general time-reversible (GTR) model of substitutions, as the best-fit model (that is, the transversion model) is not implemented in MrBayes50. Convergence of the chains was accepted when all the parameters had a sample size of at least 100 in the combined run as implemented in Tracer. To generate the consensus tree, the first 10% of generations were discarded as burn-in.
The most likely genome of the most recent common ancestor of the MTBC strains included in this study was reconstructed on the basis of the alignments of the 22 genome assemblies obtained from MAQ. Marginal and joint maximum likelihood reconstruction of the ancestral genome were obtained using the program BASEML from the PAML51 package, assuming a Jukes-Cantor model of evolution. Because the divergence between the sequences was so low, there were no undetermined states in the ancestral genome, and no difference was detected between the joint and ancestral reconstruction approaches. We used the phylogeny obtained from the SNP analysis for this purpose, specifying M. canettii as the outgroup (Fig. 1).
Genetic diversity.
Gene-by-gene genetic diversity was obtained using Variscan52, and the medians of each data set were compared by nonparametric Mann-Whitney U test using STATA s.e.m. version 10.
Calculation of dN/dS.
Because of the low number of SNPs, a gene-by-gene analysis in MTBC is uninformative. Consequently, for each category (essential, nonessential and antigens) we generated a concatenated alignment combining all individual genes. For each concatenate, we performed pairwise comparisons with the inferred ancestral genome to define synonymous and nonsynonymous substitutions, using SNAP53 and implementing the Nei-Gojobori method. Epitopes were mapped to the antigen concatenate using a FASTA search. Manual mapping was used when a particular epitope had more than one equally likely hit (for example, with antigen genes belonging to the same paralog family). Many epitopes in IEDB overlap with other epitopes, and the final set of 491 epitopes corresponded to 130 nonoverlapping regions in the antigen alignment. These nonoverlapping regions were extracted and analyzed as an epitope concatenate.
We used two different methods (measures A and B) to compare the distribution of synonymous or nonsynonymous changes across the different gene sets. In measure A, we calculated, for each concatenate, the number of nonredundant synonymous and nonsynonymous substitutions by mapping the corresponding positions onto the MTBC phylogeny. We therefore avoided counting more than once the substitutions occurring on the inner branches of the phylogeny. From the total count of nonredundant substitutions, we obtained a dN/dS ratio. The proportions of nonredundant positions that were variable at nonsynonymous and at synonymous sites were compared using a χ2 test.
In measure B, we calculated an alternative dN/dS ratio by comparing each strain to the inferred MTBC ancestor. The median dN/dS and its range across the 21 comparisons was then determined. We used this measure to test differences between essential, nonessential and antigens only. Calculation of individual strain-specific dN/dS for epitopes was not possible owing to absence of either synonymous or nonsynonymous substitutions in some of the strains. A nonparametric Mann-Whitney U test was used to compare the median dN/dS.
Accession codes.
The sequencing reads have been submitted to the NCBI Sequence Read Archive with accession codes SRX002001–SRX002005, SRX002429, SRX003589, SRX003590, SRX005394, SRX007715, SRX007716, SRX007718–SRX007726 and SRX012272. Sequence and SNP data are also available at the Tuberculosis Database.
References
Global tuberculosis control-surveillance, planning, financing (Report no. WHO/HTM/TB/2008.393). (World Health Organization, Geneva, 2009).
Shenoi, S. & Friedland, G. Extensively drug-resistant tuberculosis: a new face to an old pathogen. Annu. Rev. Med. 60, 307–320 (2009).
Hershberg, R. et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. 6, e311 (2008).
Baker, L., Brown, T., Maiden, M.C. & Drobniewski, F. Silent nucleotide polymorphisms and a phylogeny for Mycobacterium tuberculosis. Emerg. Infect. Dis. 10, 1568–1577 (2004).
Filliol, I. et al. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J. Bacteriol. 188, 759–772 (2006).
Gutacker, M.M. et al. Single-nucleotide polymorphism-based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. J. Infect. Dis. 193, 121–128 (2006).
Gagneux, S. et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 103, 2869–2873 (2006).
Gagneux, S. & Small, P.M. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect. Dis. 7, 328–337 (2007).
Reed, M.B. et al. Major Mycobacterium tuberculosis lineages associate with patient country of origin. J. Clin. Microbiol. 47, 1119–1128 (2009).
Comas, I. & Gagneux, S. The past and future of tuberculosis research. PLoS Pathog. 5, e1000600 (2009).
Woolhouse, M.E., Webster, J.P., Domingo, E., Charlesworth, B. & Levin, B.R. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat. Genet. 32, 569–577 (2002).
Brunham, R.C., Plummer, F.A. & Stephens, R.S. Bacterial antigenic variation, host immune response, and pathogen-host coevolution. Infect. Immun. 61, 2273–2276 (1993).
Dawkins, R. & Krebs, J.R. Arms races between and within species. Proc. R. Soc. Lond. B 205, 489–511 (1979).
Kawashima, Y. et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458, 641–645 (2009).
Farci, P. et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288, 339–344 (2000).
Jeffares, D.C. et al. Genome variation and evolution of the malaria parasite Plasmodium falciparum. Nat. Genet. 39, 120–125 (2007).
Urwin, R. et al. Distribution of surface protein variants among hyperinvasive meningococci: implications for vaccine design. Infect. Immun. 72, 5955–5962 (2004).
North, R.J. & Jung, Y.J. Immunity to tuberculosis. Annu. Rev. Immunol. 22, 599–623 (2004).
Shafer, R.W. & Edlin, B.R. Tuberculosis in patients infected with human immunodeficiency virus: perspective on the past decade. Clin. Infect. Dis. 22, 683–704 (1996).
Lewinsohn, D.A. et al. Immunodominant tuberculosis CD8 antigens preferentially restricted by HLA-B. PLoS Pathog. 3, 1240–1249 (2007).
Bruns, H. et al. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against Mycobacterium tuberculosis in humans. J. Clin. Invest. 119, 1167–1177 (2009).
Gutierrez, C. et al. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 1, e5 (2005).
Cole, S.T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 (1998).
Brennan, M.J. & Delogu, G. The PE multigene family: a 'molecular mantra' for mycobacteria. Trends Microbiol. 10, 246–249 (2002).
Supply, P. et al. Linkage disequilibrium between minisatellite loci supports clonal evolution of Mycobacterium tuberculosis in a high tuberculosis incidence area. Mol. Microbiol. 47, 529–538 (2003).
Ernst, J.D. et al. Meeting report: NIH workshop on the tuberculosis immune epitope database. Tuberculosis (Edinb) 88, 366–370 (2008).
Sassetti, C.M., Boyd, D.H. & Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 (2003).
Sassetti, C.M. & Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 100, 12989–12994 (2003).
Jordan, I.K., Rogozin, I.B., Wolf, Y.I. & Koonin, E.V. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 12, 962–968 (2002).
Plotkin, J.B., Dushoff, J. & Fraser, H.B. Detecting selection using a single genome sequence of M. tuberculosis and P. falciparum. Nature 428, 942–945 (2004).
Frank, S.A. & Bush, R.M. Barriers to antigenic escape by pathogens: trade-off between reproductive rate and antigenic mutability. BMC Evol. Biol. 7, 229 (2007).
Small, P.M. et al. Exogenous reinfection with multidrug-resistant Mycobacterium tuberculosis in patients with advanced HIV infection. N. Engl. J. Med. 328, 1137–1144 (1993).
Warren, R.M. et al. Patients with active tuberculosis often have different strains in the same sputum specimen. Am. J. Respir. Crit. Care Med. 169, 610–614 (2004).
Daley, C.L. et al. An outbreak of tuberculosis with accelerated progression among persons infected with the human immunodeficiency virus. An analysis using restriction-fragment-length polymorphisms. N. Engl. J. Med. 326, 231–235 (1992).
Blaser, M.J. & Kirschner, D. The equilibria that allow bacterial persistence in human hosts. Nature 449, 843–849 (2007).
Rodrigo, T. et al. Characteristics of tuberculosis patients who generate secondary cases. Int. J. Tuberc. Lung Dis. 1, 352–357 (1997).
Mukadi, Y. et al. Spectrum of immunodeficiency in HIV-1-infected patients with pulmonary tuberculosis in Zaire. Lancet 342, 143–146 (1993).
Fraser, C., Hollingsworth, T.D., Chapman, R., de Wolf, F. & Hanage, W.P. Variation in HIV-1 set-point viral load: epidemiological analysis and an evolutionary hypothesis. Proc. Natl. Acad. Sci. USA 104, 17441–17446 (2007).
Young, D.B., Perkins, M.D., Duncan, K. & Barry, C.E. Confronting the scientific obstacles to global control of tuberculosis. J. Clin. Invest. 118, 1255–1265 (2008).
Andersen, P. & Doherty, T.M. Opinion: the success and failure of BCG—implications for a novel tuberculosis vaccine. Nat. Rev. Microbiol. 3, 656–662 (2005).
Colditz, G.A. et al. Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. J. Am. Med. Assoc. 271, 698–702 (1994).
Pizza, M. et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 287, 1816–1820 (2000).
Abdallah, A.M. et al. Type VII secretion–mycobacteria show the way. Nat. Rev. Microbiol. 5, 883–891 (2007).
Rocha, E.P. et al. Comparisons of dN/dS are time dependent for closely related bacterial genomes. J. Theor. Biol. 239, 226–235 (2006).
Holt, K.E. et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet. 40, 987–993 (2008).
Li, H., Ruan, J. & Durbin, R. Mapping short DNA sequencing reads and callin variants using mapping quality scores. Genome Res. 18, 1851–1858 (2008).
Tamura, K., Dudley, J., Nei, M. & Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599 (2007).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Posada, D. & Crandall, K.A. MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Ronquist, F. & Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Vilella, A.J., Blanco-Garcia, A., Hutter, S. & Rozas, J. VariScan: Analysis of evolutionary patterns from large-scale DNA sequence polymorphism data. Bioinformatics 21, 2791–2793 (2005).
Korber, B. Computational Analysis of HIV Molecular Sequences (Kluwer Academic Publishers, Dordrecht, The Netherlands, 2000).
Acknowledgements
We thank F. Gonzalez-Candelas, S. Borrell and D. Young for comments on the manuscript. This project has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Disease, US National Institutes of Health (NIH), US Department of Health and Human Services, under contract no. HHSN266200400001C. J.C. is a Howard Hughes Medical Institute Research Training Fellow. J.D.E. was supported by NIH grants AI046097 and AI051242 and S.G. by the Medical Research Council UK, the Royal Society, the Swiss National Science Foundation and NIH grants HHSN266200700022C and AI034238.
Author information
Authors and Affiliations
Contributions
I.C., J.D.E. and S.G. designed the study; P.M.S., S.N., K.K. and S.G. contributed sources of M. tuberculosis DNA and demographic information; I.C., J.C. and J.G. performed DNA sequencing and bioinformatics; I.C., P.M.S., J.D.E. and S.G. wrote the manuscript with comments from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1 and 2 and Supplementary Tables 1–6. (PDF 735 kb)
Rights and permissions
About this article
Cite this article
Comas, I., Chakravartti, J., Small, P. et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet 42, 498–503 (2010). https://doi.org/10.1038/ng.590
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.590
This article is cited by
-
Genotypic and phenotypic comparison of drug resistance profiles of clinical multidrug-resistant Mycobacterium tuberculosis isolates using whole genome sequencing in Latvia
BMC Infectious Diseases (2023)
-
Helper T cell bias following tuberculosis chemotherapy identifies opportunities for therapeutic vaccination to prevent relapse
npj Vaccines (2023)
-
Attenuated but immunostimulatory Mycobacterium tuberculosis variant bovis strain Ravenel shows variation in T cell epitopes
Scientific Reports (2023)
-
A smooth tubercle bacillus from Ethiopia phylogenetically close to the Mycobacterium tuberculosis complex
Nature Communications (2023)
-
T cell receptor repertoires associated with control and disease progression following Mycobacterium tuberculosis infection
Nature Medicine (2023)
