
Abstract
Brain malformations involving the corpus callosum are common in children with developmental disabilities. We identified DCC mutations in four families and five sporadic individuals with isolated agenesis of the corpus callosum (ACC) without intellectual disability. DCC mutations result in variable dominant phenotypes with decreased penetrance, including mirror movements and ACC associated with a favorable developmental prognosis. Possible phenotypic modifiers include the type and location of mutation and the sex of the individual.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others
References
Lindwall, C., Fothergill, T. & Richards, L.J. Curr. Opin. Neurobiol. 17, 3–14 (2007).
Chédotal, A. Curr. Opin. Neurobiol. 21, 68–75 (2011).
Paul, L.K. et al. Nat. Rev. Neurosci. 8, 287–299 (2007).
Glass, H.C., Shaw, G.M., Ma, C. & Sherr, E.H. Am. J. Med. Genet. A. 146A, 2495–2500 (2008).
Rouleau, C. et al. Arch. Dis. Child. Fetal Neonatal Ed. 96, F360–F364 (2011).
Edwards, T.J., Sherr, E.H., Barkovich, A.J. & Richards, L.J. Brain 137, 1579–1613 (2014).
Sotiriadis, A. & Makrydimas, G. Am. J. Obstet. Gynecol. 206, 337.e1–337.e5 (2012).
Fazeli, A. et al. Nature 386, 796–804 (1997).
Srour, M. et al. Science 328, 592 (2010).
Méneret, A. et al. Neurology 82, 1999–2002 (2014).
Ardekani, B.A., Figarsky, K. & Sidtis, J.J. Cereb. Cortex 23, 2514–2520 (2013).
Moffat, S.D., Hampson, E., Wickett, J.C., Vernon, P.A. & Lee, D.H. Brain Res. 767, 297–304 (1997).
Chura, L.R. et al. Psychoneuroendocrinology 35, 122–132 (2010).
Xu, K. et al. Science 344, 1275–1279 (2014).
Greig, L.C., Woodworth, M.B., Galazo, M.J., Padmanabhan, H. & Macklis, J.D. Nat. Rev. Neurosci. 14, 755–769 (2013).
Finci, L.I. et al. Neuron 83, 839–849 (2014).
Fothergill, T. et al. Cereb. Cortex 24, 1138–1151 (2014).
Gallea, C. et al. Brain 136, 3333–3346 (2013).
Andersson, J.L. & Sotiropoulos, S.N. Neuroimage 125, 1063–1078 (2016).
Andersson, J.L., Skare, S. & Ashburner, J. Neuroimage 20, 870–888 (2003).
Smith, S.M. et al. Neuroimage 23 (Suppl. 1), S208–S219 (2004).
Tournier, J.D., Calamante, F., Gadian, D.G. & Connelly, A. Neuroimage 23, 1176–1185 (2004).
Tournier, J.D., Calamante, F. & Connelly, A. Neuroimage 35, 1459–1472 (2007).
Tournier, J.D., Calamante, F. & Connelly, A. Int. J. Imaging Syst. Technol. 22, 53–66 (2012).
Li, H. & Durbin, R. Bioinformatics 26, 589–595 (2010).
McKenna, A. et al. Genome Res. 20, 1297–1303 (2010).
Smith, K.R. et al. Genome Biol. 12, R85 (2011).
Depienne, C. et al. Neurology 76, 260–264 (2011).
Dupasquier, S. et al. BMC Mol. Biol. 15, 9 (2014).
Lauck, F., Smith, C.A., Friedland, G.F., Humphris, E.L. & Kortemme, T. Nucleic Acids Res. 38, W569–W575 (2010).
Pettersen, E.F. et al. J. Comput. Chem. 25, 1605–1612 (2004).
Dolinsky, T.J., Nielsen, J.E., McCammon, J.A. & Baker, N.A. Nucleic Acids Res. 32, W665–W667 (2004).
Baker, N.A., Sept, D., Joseph, S., Holst, M.J. & McCammon, J.A. Proc. Natl. Acad. Sci. USA 98, 10037–10041 (2001).
Laskowski, R.A. & Swindells, M.B. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Landau, M. et al. Nucleic Acids Res. 33, W299–W302 (2005).
Acknowledgements
We thank the studied families and the Lefroy family for their participation in and support of this study. We thank the DNA and cell bank of the ICM (Paris, France) for DNA extraction, Sinead Eyre (QBI) for study coordination and M. Kean (RCH) and M. Seal (MCRI) for assistance with MRI protocols and scanning. This work was funded in part by National Health and Medical Research Council (NHMRC) Australia Project grants (GNT1059666, GNT631466, GNT1064174, GNT1048849, GNT1104455 and GNT1064174), the Agence Nationale de la Recherche (ANR Blanc CILAXCAL, ANR Blanc HARTaGeNe), Assistance Publique des Hôpitaux de Paris (APHP), the 'Programme Hospitalier de Recherche Clinique' (PHRC) ACCREM, and the 'Investissements d'Avenir' programs ANR-10-IAIHU-06 (IHU-A-ICM), ANR-10-LABX-0030-INRT and ANR-10-IDEX-0002-02. A.P.L.M. and L.R.M. are supported by an Australian Postgraduate Award, T.J.E. is supported by a University of Queensland Research Scholarship, and A.P. is supported by a QBI PhD scholarship. S.H. and A.Q. are respectively supported by a master's and a doctoral grant from the Fondation pour la Recherche Médicale (FRM). M.B. is supported by an NHMRC Senior Research Fellowship and an NHMRC Program Grant (GNT1054618). E.H.S. is supported by a grant from the NIH (2R01NS058721), and R.J.L. is supported by a Melbourne Children's Clinician Scientist Fellowship. L.J.R. is supported by an NMHRC Principal Research Fellowship, and P.J.L. is supported by an NHMRC Career Development Fellowship (GNT1032364). C.D. and C.N. are supported as members of the Bio-Psy Labex. This work was supported in part by the Victorian Government's Operational Infrastructure Support Program and Australian Government NHMRC IRIISS.
Author information
Authors and Affiliations
Contributions
A.P.L.M., D.H., T.J.E., C. Galea, E.H.S., R.J.L., L.J.R., P.J.L. and C.D. contributed to formulation of theory and prediction. A.P.L.M., D.H., T.J.E., C.N., S.E.M.S., J.-L.M., A. Piton, L.J.R., P.J.L. and C.D. contributed to experimental conception and design.A.P.L.M., D.H., T.J.E., C. Galea, A.Q., C.N., A.R., M.-L.M., V.A., P.B., J.B., A.F., C. Garel, G.G., I.G., J.G., S.H., B.K., F.L., V.L., S.A.M., G.M., A. McIlroy, A. Meneret, C.M., L.R.M., S.O., A. Paolino, K.P., F.R., G.A.R., M.S.-S., M.S., S.E.M.S., R.T., O.T., Q.W., A.W., E.R., A. Piton, M.B., T.B.d.V., E.H.S., R.J.L., L.J.R., P.J.L. and C.D. contributed to acquisition, analysis and/or interpretation of data. A.P.L.M., T.J.E., P.J.L. and C.D. contributed to drafting the article. A.P.L.M., D.H., T.J.E., A.Q., C. Galea, C.N., M.-L.M., V.A., S.A.M., G.M., A.M., G.A.R., A.B., G.R., T.A.-B., M.B.D., J.-L.M., D.J.A., E.R., A. Piton, M.B., T.B.d.V., E.H.S., R.J.L., L.J.R., P.J.L. and C.D. contributed to critically revising the article for important intellectual content.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
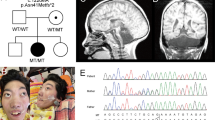
Supplementary Figure 1 Pedigrees and MRI of sporadic DCC-mutant individuals.
(a) Pedigrees of sporadic individuals 5, 6, 7, 8 and 9. The proband of each pedigree are indicated by a black arrowhead; * indicates neuroimaging for mutation carrier. (b) Midsagittal MRI of probands 5-9 showing complete ACC in all individuals. Proband 9 has an intrahemispheric cyst in addition to ACC. (c) Midsagittal MRI of additional members of families 2 and 4 with DCC mutation showing complete isolated ACC in affected individuals of family 2 and the presence of a normal corpus callosum in both individuals with MMs only in family 4.
Supplementary Figure 2 Mutations in DCC are associated with decreased crossing of descending corticospinal tract projections at the pyramidal decussation.
(a) Upper panel: Color-coded crossed (orange and blue) and uncrossed (purple and yellow) corticospinal tracts, and the bilateral regions of interest used to reconstruct these tracts at: the base of the pons (i), the pyramid of the upper medulla (ii), the pyramid of the lower medulla (iii) and the lateral funiculus of the cervical cord (iv). Lower panel: Regions of interest projected over the color fractional anisotropy map. (b) Reconstruction of the corticospinal tracts by tractography demonstrates reduced crossed (orange and blue) and more uncrossed streamlines (purple and yellow) in DCC mutant individuals with MMs with and without ACC (representative images, II-1 of family 2 and III-1 of family 4) compared to controls.
Supplementary Figure 3 LOD scores obtained from parametric linkage analysis of family 1 under a dominant genetic model.
LOD scores are on the y-axis and cumulative genomic position in Mb on the x-axis. The vertical lines depict the boundaries of each chromosome. The peak LOD score of 1.8 is achieved at 16 sites on chromosomes 1, 2, 3, 7, 8, 11, 15, and 18.
Supplementary Figure 4 LOD scores obtained from parametric linkage analysis of family 2 under a dominant genetic model.
LOD scores are on the y-axis and cumulative genomic position in cM on the x-axis. The vertical lines depict the boundaries of each chromosome. The peak LOD score of 0.6 is achieved at 28 sites.
Supplementary Figure 6 Sex-biased phenotypic expression of ACC and MMs in individuals with truncating DCC mutations may be associated with regulation of DCC by testosterone.
(a) DCC mRNA were quantified by RT-qPCR in NSCs treated with DMSO (n = 14), 10 nM (n = 12) or 100 nM testosterone (n = 12) for 24 h using two different primer pairs, located in exons 9-10 (DCC_F1R1, above) and 1-2 (DCC_F2R2, below) of DCC, respectively, and two different control genes (Left: GAPDH; Right: PPIA). Each condition includes the values calculated from three independent experiments each composed of at least 4 well replicates (6 replicates for the DMSO condition in experiment 1) and quantification was performed in triplicates. The relative expression of DCC (mean with range) versus GAPDH or PPIA was compared using a two-sided parametric Student’s t-test, assuming equal variance (degree of freedom = N1 + N2-2); significant P-values are indicated by the * for 100 nM testosterone treatment (DCC_F1R1_GAPDH: P = 0.0127; DCC_F2R2_GAPDH: P = 0.0423; DCC_F1R1_PIAA: P = 0.0127; DCC_F2R2_PIAA: p=0.0152). Numbers in red indicate the mean ratio induction (the mean for the DMSO condition is equal to 1). (b) Combined analysis comparing the mean value (with range) of DCC expression in cells treated with DMSO (n = 52), testosterone 10 nM (n = 46) or testosterone 100 nM (n = 48). P-values: **P = 0.0038; ****P = 1.46 x 10-6.
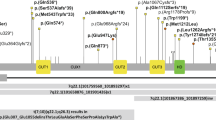
Supplementary Figure 7 DCC missense variants in the DCC/Netrin-1 binding interface are predicted to be highly disruptive.
(a) Structural model with DCC the orange ribbon and Netrin-1 as a white solvent accessible surface, mutations are represented as blue spheres. (b) Expansion of the binding interface (red dotted box) with Netrin-1 residues colored blue and critical DCC residues represented as purple sticks. LN, laminin domain; LE, laminin-type EGF-like domain; FN, fibronectin type III-like domain. Protein Data Bank ID: 4PLO.
Supplementary Figure 8 The DCC/Netrin-1 binding-interface substitutions p.Val793Gly and p.Gly805Glu.
(a) Conservation of residues on the surface of the DCC FN4 domain determined using the program ConSurf where conserved residues are colored magenta, residues of average conservation are white and variable amino acids are cyan. (b,c) Ribbon representation of the DCC FN4 domain where wild-type and mutated residues for p.(Val793Gly) and p.(Gly805Glu) are represented as grey sticks and outlined by a black dotted box. Insets show expanded regions containing the wild-type and mutant residues.
Supplementary Figure 9 Binding of Netrin-1 to DCC FN4 and FN5 domains.
(a) Structure of Netrin-1 is represented by a translucent white molecular surface while DCC is an orange ribbon. FN4, fibronectin type III-like domain 4; FN5, fibronectin type III-like domain 5. Protein Data Bank ID: 4URT. (b) Expansion of red dotted box region in a showing residues of Netrin-1 (magenta sticks) and DCC (white sticks) that are involved in binding. Hydrogen bonds are represented as dotted black lines and water molecule as a red ball.
Supplementary Figure 10 The ACC-associated Netrin-1-binding-region p.Met743Leu mutation.
(a) Structure of the DCC/Netrin-1 complex where DCC is shown as an orange ribbon and Netrin-1 as a white solvent accessible surface. The region of Netrin-1 that binds to DCC is colored blue. FN4, fibronectin type III-like domain 4; FN5, fibronectin type III-like domain 5. (b) Expansion of the red dotted box region containing the M743 residue (magenta sticks) overlaid onto the p.Met743Leu mutant (green sticks). Conserved residues close to the mutated residue M743 are shown as light blue sticks. M743 is highly conserved and interacts with several predominately hydrophobic residues (light blue sticks) within the core of the protein that are also highly conserved.
Supplementary Figure 11 The ACC-associated Netrin-1-binding-region p.Val754Met mutation.
(a) Structure of the DCC/Netrin-1 complex where DCC is shown as an orange ribbon and Netrin-1 as a white solvent accessible surface. The region of Netrin-1 that binds to DCC is colored blue. FN4, fibronectin type III-like domain 4; FN5, fibronectin type III-like domain 5. (b) Expansion of the region containing the V754 residue (magenta sticks) overlaid onto the p.Val754Met mutant (green sticks). Hydrogen bond is depicted by a dotted black line. Residue V754 lies in a loop on the surface of the protein that connects β-stands from each of the two DCC β-sheets and interacts with residue E722 within the N-terminal tail of the DCC FN4 domain.
Supplementary Figure 12 The ACC-associated Netrin-1-binding-region p.Ala893Thr mutation.
(a) Structure of Netrin-1 (white solvent accessible surface) bound to DCC, FN5 and FN6 domains (orange ribbons). The region of Netrin-1 that binds to DCC is colored blue. FN5, fibronectin type III-like domain 5; FN6, fibronectin type III-like domain 6, Protein Data Bank ID: 4URT. (b) Expansion of the region containing the A893 residue (white sticks) and p.Ala893Thr mutation (green sticks). A893 lies within the DCC FN5 domain, which has also been shown to bind to Netrin-1.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–12 and Supplementary Tables 1–7 (PDF 3958 kb)
Rights and permissions
About this article

Cite this article
Marsh, A., Heron, D., Edwards, T. et al. Mutations in DCC cause isolated agenesis of the corpus callosum with incomplete penetrance. Nat Genet 49, 511–514 (2017). https://doi.org/10.1038/ng.3794
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3794
This article is cited by
-
Corticolimbic DCC gene co-expression networks as predictors of impulsivity in children
Molecular Psychiatry (2022)
-
Penetrance estimation of PRRT2 variants in paroxysmal kinesigenic dyskinesia and infantile convulsions
Frontiers of Medicine (2021)
-
Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease
Human Genetics (2021)
-
X-linked partial corpus callosum agenesis with mild intellectual disability: identification of a novel L1CAM pathogenic variant
neurogenetics (2021)
-
25-jähriger Altenpfleger mit Koordinationsstörungen der oberen Extremitäten
DGNeurologie (2020)
