
Abstract
Bosma arhinia microphthalmia syndrome (BAMS) is an extremely rare and striking condition characterized by complete absence of the nose with or without ocular defects. We report here that missense mutations in the epigenetic regulator SMCHD1 mapping to the extended ATPase domain of the encoded protein cause BAMS in all 14 cases studied. All mutations were de novo where parental DNA was available. Biochemical tests and in vivo assays in Xenopus laevis embryos suggest that these mutations may behave as gain-of-function alleles. This finding is in contrast to the loss-of-function mutations in SMCHD1 that have been associated with facioscapulohumeral muscular dystrophy (FSHD) type 2. Our results establish SMCHD1 as a key player in nasal development and provide biochemical insight into its enzymatic function that may be exploited for development of therapeutics for FSHD.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Accession codes
References
Brasseur, B., Martin, C.M., Cayci, Z., Burmeister, L. & Schimmenti, L.A. Bosma arhinia microphthalmia syndrome: clinical report and review of the literature. Am. J. Med. Genet. A. 170A, 1302–1307 (2016).
Graham, J.M. Jr. & Lee, J. Bosma arhinia microphthalmia syndrome. Am. J. Med. Genet. A. 140, 189–193 (2006).
Shaw, N.D. et al. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet. http://dx.doi.org/10.1038/ng.3743 (2017).
Forni, P.E. & Wray, S. GnRH, anosmia and hypogonadotropic hypogonadism—where are we? Front. Neuroendocrinol. 36, 165–177 (2015).
Szabo-Rogers, H.L. et al. Novel skeletogenic patterning roles for the olfactory pit. Development 136, 219–229 (2009).
Blewitt, M.E. et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 40, 663–669 (2008).
Nickell, M.D., Breheny, P., Stromberg, A.J. & McClintock, T.S. Genomics of mature and immature olfactory sensory neurons. J. Comp. Neurol. 520, 2608–2629 (2012).
Tryggestad, J.B., Li, S. & Chernausek, S.D. Hypogonadotropic hypogonadism presenting with arhinia: a case report. J. Med. Case Rep. 7, 52 (2013).
Gendrel, A.-V. et al. Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev. Cell 23, 265–279 (2012).
Gendrel, A.-V. et al. Epigenetic functions of Smchd1 repress gene clusters on the inactive X chromosome and on autosomes. Mol. Cell. Biol. 33, 3150–3165 (2013).
Nozawa, R.-S. et al. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1–HBiX1 pathway. Nat. Struct. Mol. Biol. 20, 566–573 (2013).
Mould, A.W. et al. Smchd1 regulates a subset of autosomal genes subject to monoallelic expression in addition to being critical for X inactivation. Epigenetics Chromatin 6, 19 (2013).
Chen, K. et al. Genome-wide binding and mechanistic analyses of Smchd1-mediated epigenetic regulation. Proc. Natl. Acad. Sci. USA 112, E3535–E3544 (2015).
Coker, H. & Brockdorff, N. SMCHD1 accumulates at DNA damage sites and facilitates the repair of DNA double-strand breaks. J. Cell Sci. 127, 1869–1874 (2014).
Tang, M. et al. Structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) promotes non-homologous end joining and inhibits homologous recombination repair upon DNA damage. J. Biol. Chem. 289, 34024–34032 (2014).
Lemmers, R.J.L.F. et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 44, 1370–1374 (2012).
Hewitt, J.E. Loss of epigenetic silencing of the DUX4 transcription factor gene in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 24 R1, R17–R23 (2015).
Lemmers, R.J.L.F. et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329, 1650–1653 (2010).
Sacconi, S. et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am. J. Hum. Genet. 93, 744–751 (2013).
Lemmers, R.J.L.F. et al. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 24, 659–669 (2015).
de Greef, J.C. et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 75, 1548–1554 (2010).
Chen, K. et al. The epigenetic regulator Smchd1 contains a functional GHKL-type ATPase domain. Biochem. J. 473, 1733–1744 (2016).
Brideau, N.J. et al. Independent mechanisms target SMCHD1 to trimethylated histone H3 lysine 9–modified chromatin and the inactive X chromosome. Mol. Cell. Biol. 35, 4053–4068 (2015).
Dutta, R. & Inouye, M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 25, 24–28 (2000).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Flicek, P. et al. Ensembl 2013. Nucleic Acids Res. 41, D48–D55 (2013).
DePristo, M.A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Van der Auwera, G.A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013).
Javed, A., Agrawal, S. & Ng, P.C. Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat. Methods 11, 935–937 (2014).
Sherry, S.T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 (2001).
Mishima, H., Sasaki, K., Tanaka, M., Tatebe, O. & Yoshiura, K. Agile parallel bioinformatics workflow management using Pwrake. BMC Res. Notes 4, 331 (2011).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Adzhubei, I.A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Kumar, P., Henikoff, S. & Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899 (2011).
Liu, X., Wu, C., Li, C. & Boerwinkle, E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 37, 235–241 (2016).
Magdinier, F. et al. Regional methylation of the 5′ end CpG island of BRCA1 is associated with reduced gene expression in human somatic cells. FASEB J. 14, 1585–1594 (2000).
Li, L.-C. & Dahiya, R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 18, 1427–1431 (2002).
Bock, C. et al. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 21, 4067–4068 (2005).
Gaillard, M.-C. et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology 83, 733–742 (2014).
Gaillard, M.-C. et al. Segregation between SMCHD1 mutation, D4Z4 hypomethylation and facio-scapulo-humeral dystrophy: a case report. BMC Med. Genet. 17, 66 (2016).
Kelley, L.A., Mezulis, S., Yates, C.M., Wass, M.N. & Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Thompson, J.D., Higgins, D.G. & Gibson, T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Rosin, N. et al. Mutations in XRCC4 cause primary microcephaly, short stature and increased genomic instability. Hum. Mol. Genet. 24, 3708–3717 (2015).
Verkaik, N.S. et al. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol. 32, 701–709 (2002).
Acknowledgements
We would like to thank all family members and their relatives for their participation and kind contribution to this study. N. Akarsu was instrumental for recruiting patient 11. Support from the Jean Renny Endowed Chair for Craniofacial Research (M.L.C.) is acknowledged. C.D. is the recipient of a fellowship from the French Ministry of Education and Research. H.F. was supported by a postdoctoral grant from INSERM. B.R. is a fellow of the Branco Weiss Foundation, an A*STAR Investigator, an EMBO Young Investigator and a recipient of the inaugural AAA Fellowship in Amsterdam. This work was supported by funding from the Agence Nationale de la Recherche (ANR-10-IAHU-01, CranioRespiro), the Cancer Council Victoria (fellowship to K.C.), the National Health and Medical Research Council (NHMRC) of Australia to M.E.B. and J.M.M. (1098290 and fellowships 1110206 and 1105754), the Scientific and Technological Research Council of Turkey (TUBITAK) to H.K. (grant 112S398, E-RARE network CRANIRARE-2), the Association Française contre les Myopathies (AFM) to F.M., Victorian State Government Operational Infrastructure Support, an NHMRC IRIISS grant (9000220), the German Federal Ministry of Education and Research (BMBF) to B.W. (grant 01GM1211A, E-RARE network CRANIRARE-2), the German Research Foundation (SFB1002, project D02) to B.W., MACS, VICTA and Baillie Gifford grant support to N. Ragge, Mahidol University and Research Career Development Awards from the Faculty of Medicine Ramathibodi Hospital to D. Wattanasirichaigoon, an A*STAR JCO Career Development grant to A.J., an A*STAR BMRC Young Investigator Grant to S.X. and a Strategic Positioning Fund on Genetic Orphan Diseases from the Biomedical Research Council, A*STAR, Singapore, to B.R.
Author information
Authors and Affiliations
Contributions
Genetic studies were performed by H.F., C.T.G., M.O., K.Y., C.B.-F., P. Nitschké, P. Nürnberg, C.B., A.S.M.T., A.J., H.T., J. Altmüller and G.Y. Genetic studies were supervised by C.T.G., J. Amiel, B.W., A.M.H. and B.R. The team consisting of B.R., A.J., S.X., H.K. and D. Wattanasirichaigoon independently identified SMCHD1 mutations in patients 9–12 and 14. H.K., D. Wattanasirichaigoon, C.C., G.T., N. Ragge, R.M., A.C.M., N.O., V.V., R.I., S.S., D. Williams, S.F.A., I.R., N.F., M.F., S.C.E., H.R., A.S., S.L., D.M., W.M. and M.L.C. diagnosed patients. K.C., A.D.G., J.M.M. and M.E.B. performed and analyzed the results of ATPase assays. S.X., M.K.K. and B.R. performed and analyzed the results of functional experiments in Xenopus. N. Rosin and G.Y. performed DNA damage repair assays, supervised by B.W. C.T.G. and T.J.B. performed analysis of Smchd1gt/+ embryos. C.D., N.L. and F.M. performed and analyzed the results of methylation studies. The manuscript was written by C.T.G. with contributions from S.X., H.F., J. Amiel and B.R. All authors read and approved its content.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
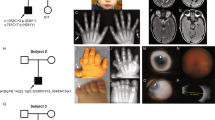
Supplementary Figure 1 Computed tomography and magnetic resonance imaging (MRI) in BAMS.
(a–f) Controls (a–c) and patient 1 (d–f) at 4 years. Patient 1 displays maxillary hypoplasia and absent nasal bones (d,e). Olfactory bulbs and sulci (labeled with red and white arrows, respectively, on the left side in c) are absent in patient 1 (f). (g–i) Patient 5, with right microphthalmia, as shown by MRI (i). (j–l) Skeletal imaging of patient 14 (j,k) and patient 11 (l) indicating midface hyploplasia.
Supplementary Figure 2 BAMS pedigrees and Sanger sequencing chromatograms of SMCHD1 mutations.
Individuals submitted for exome sequencing are indicated by a red asterisk. Note that Sanger sequencing was unavailable for individual 13.
Supplementary Figure 3 Multiple-sequence alignment of vertebrate SMCHD1 orthologs and yeast Hsp90.
Residues mutated in BAMS are indicated by pink arrows. Residues mutated in FSHD are indicated by purple arrows. The conserved GHKL-ATPase motifs I–IV are shaded in blue. Hs, Homo sapiens; Mm, Mus musculus; Bt, Bos taurus; Gg, Gallus gallus; Md, Monodelphis domestica; Cm, Chelonia mydas; Xt, Xenopus tropicalis; Dr, Danio rerio; Sc, Saccharomyces cerevisiae. FSHD mutation reference: LOVD SMCHD1 variant database (see URLs).
Supplementary Figure 4 X-gal staining of mouse embryos expressing lacZ from the Smchd1 locus.
(a–e and j-q) Embryos. E, embryonic day; gt/+, embryos heterozygous for the Smchd1gt allele expressing LacZ; +/+, wild-type embryos; hf, head folds; npl, nasal placode; ov, optic vesicle; npi, nasal pit; ne, nasal epithelium. (f–i) Coronal sections. (r,s) Transverse sections. An asterisk in p indicates deep nasal staining.
Supplementary Figure 5 Sodium bisulfite sequencing in patients with BAMS (individuals 1–6).
The position of the three different regions analyzed within D4Z4 is indicated above the corresponding column (left, DR1; middle, 5′; right, Mid). For each sample, at least ten cloned DNA molecules were analyzed by Sanger sequencing. Each histogram column corresponds to a single CpG. Black corresponds to the global percentage of methylated CpGs; white corresponds to the global percentage of unmethylated CpGs. The percentage of methylated CpGs out of the total CpGs in each individual analyzed is given in Supplementary Table 3.
Supplementary Figure 6 Sodium bisulfite sequencing in patients with BAMS (individuals 8–11 and 14).
See the legend of Supplementary Figure 5 for further information.
Supplementary Figure 7 Comparison of D4Z4 methylation in patients with BAMS or FSHD2, the relatives of patients with BAMS and controls.
Distribution of methylation for the three different regions within the D4Z4 sequence (DR1, 5′ and Mid) in control individuals, patients with FSHD2 carrying an SMCHD1 mutation, and patients with BAMS and their relatives. Means ± s.e.m. are shown. A Kruskal–Wallis multiple-comparisons test was performed, followed by a Dunn’s test and Bonferroni correction, with α = 0.05. ***P < 0.0001, **P < 0.001, *P < 0.05. Blue points represent outliers; red crosses represent medians. The level of methylation is statistically significantly different between controls and patients with FSHD2 for the DR1 (**P < 0.001) and 5′ (***P < 0.0001) regions. The level of methylation is significantly different between controls and patients with BAMS for the 5′ region (*P < 0.05) and between patients with BAMS and their relatives for the DR1 (*P < 0.05) and 5′ (**P < 0.001) regions.
Supplementary Figure 8 Structural model of the N-terminal region of mouse Smchd1, based on the crystal structure of Hsp90.
Residues mutated in BAMS are shown in pink. Residues mutated in FSHD are shown in purple. Motif I is shown in blue. ATP is shown in orange.
Supplementary Figure 9 Fibroblasts derived from patients with BAMS show no defects in NHEJ or in H2AX activation.
(a) A microhomology-mediated end-joining (MMEJ) assay was performed on wild-type (WT), XRCC4-deficient and case 1 and 2 fibroblasts. Whereas XRCC4-deficient fibroblasts show multiple smaller DNA bands after BstXI digestion indicating defects in NHEJ-mediated DNA repair and leading to preferential use of MMEJ-mediated DNA double-strand repair, fibroblasts from patients with BAMS show no defects in NHEJ-mediated DNA repair pathways in comparison to wild-type fibroblasts. (b) Immunoblot analysis of UV- and etoposide-induced phosphorylation of H2AX at Ser139 (γH2AX). Wild-type fibroblasts (WT) and fibroblasts derived from cases 1 and 2 were treated with UV-C (UV) or etoposide (Eto) or left untreated as a control (–). Cells were lysed and subjected to immunoblot analysis with an antibody against γH2AX. Equal protein loading was confirmed by reprobing of the membrane with an antibody against β-actin. Wild-type fibroblasts and those from patients with BAMS did not show significant differences in H2AX activation.
Supplementary Figure 10 ATPase assays performed using wild-type or mutant recombinant mouse Smchd1 protein in the presence of radicicol.
Data are displayed as means ± s.d. from three technical replicates. The data are representative of at least two independent experiments using different batches of protein preparation.
Supplementary Figure 11 Full-length immunoblot.
Full-length immunoblot of the cropped blot image in Figure 3g.
Supplementary Figure 12 SMCHD1 overexpression in Xenopus causes dose-dependent craniofacial anomalies.
(a,b) Measurements of eye diameter of Xenopus embryos injected with 240 pg (a) or 500 pg (b) SMCHD1 mRNA. Y353C is an FSHD2 mutation. At least 20 embryos were studied for each condition. (c–f) Representative Xenopus embryos injected with 500 pg of wild-type or FSHD2 mutant SMCHD1 or 120 pg of BAMS mutant mRNA show varying degrees of craniofacial abnormalities as compared to uninjected control tadpoles at 4 days post-fertilization. Data are shown as means ± s.d. P values were calculated by Kruskal–Wallis test followed by Dunn’s post test. n.s., not significant.
Supplementary Figure 13 Purity of proteins used for ATPase assays.
Purified recombinant wild-type or mutant proteins were resolved by 4–20% Tris-glycine reducing SDS–PAGE and were stained with SimplyBlue SafeStain. The protein quantities loaded were as follows: left gel, 1.4 μg; middle gel, 1.05 μg; right gel, 0.7 μg. The sizes of molecular weight (MW) markers are as indicated on the left-hand side.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–13 and Supplementary Tables 3–6 (PDF 3070 kb)
Supplementary Table 1
Clinical features of 14 patients with BAMS. (XLSX 13 kb)
Supplementary Table 2
Exome variant filtering for cases 1, 2 and 9–13. (XLSX 11 kb)
Rights and permissions
About this article

Cite this article
Gordon, C., Xue, S., Yigit, G. et al. De novo mutations in SMCHD1 cause Bosma arhinia microphthalmia syndrome and abrogate nasal development. Nat Genet 49, 249–255 (2017). https://doi.org/10.1038/ng.3765
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3765
This article is cited by
-
Cholesterol biosynthesis modulates differentiation in murine cranial neural crest cells
Scientific Reports (2023)
-
SMCHD1 and LRIF1 converge at the FSHD-associated D4Z4 repeat and LRIF1 promoter yet display different modes of action
Communications Biology (2023)
-
Facioscapulohumeral muscular dystrophy: the road to targeted therapies
Nature Reviews Neurology (2023)
-
SMCHD1 has separable roles in chromatin architecture and gene silencing that could be targeted in disease
Nature Communications (2023)
-
Meeting report: the 2021 FSHD International Research Congress
Skeletal Muscle (2022)
