
Abstract
Insulin resistance is a key mediator of obesity-related cardiometabolic disease, yet the mechanisms underlying this link remain obscure. Using an integrative genomic approach, we identify 53 genomic regions associated with insulin resistance phenotypes (higher fasting insulin levels adjusted for BMI, lower HDL cholesterol levels and higher triglyceride levels) and provide evidence that their link with higher cardiometabolic risk is underpinned by an association with lower adipose mass in peripheral compartments. Using these 53 loci, we show a polygenic contribution to familial partial lipodystrophy type 1, a severe form of insulin resistance, and highlight shared molecular mechanisms in common/mild and rare/severe insulin resistance. Population-level genetic analyses combined with experiments in cellular models implicate CCDC92, DNAH10 and L3MBTL3 as previously unrecognized molecules influencing adipocyte differentiation. Our findings support the notion that limited storage capacity of peripheral adipose tissue is an important etiological component in insulin-resistant cardiometabolic disease and highlight genes and mechanisms underpinning this link.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Change history
05 December 2016
In the version of this article initially published online, the middle initial of collaborator Maarten R. Soeters was inadvertently omitted. The error has been corrected for the print, PDF and HTML versions of this article.
References
Samuel, V.T. & Shulman, G.I. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest. 126, 12–22 (2016).
Ginsberg, H.N. Insulin resistance and cardiovascular disease. J. Clin. Invest. 106, 453–458 (2000).
Shulman, G.I. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl. J. Med. 371, 1131–1141 (2014).
Lillioja, S. & Bogardus, C. Obesity and insulin resistance: lessons learned from the Pima Indians. Diabetes Metab. Rev. 4, 517–540 (1988).
Perry, R.J., Samuel, V.T., Petersen, K.F. & Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510, 84–91 (2014).
Arner, P. The adipocyte in insulin resistance: key molecules and the impact of the thiazolidinediones. Trends Endocrinol. Metab. 14, 137–145 (2003).
Friedman, J.M. Obesity in the new millennium. Nature 404, 632–634 (2000).
Guilherme, A., Virbasius, J.V., Puri, V. & Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 9, 367–377 (2008).
Friedman, J.M. A war on obesity, not the obese. Science 299, 856–858 (2003).
Hardy, O.T., Czech, M.P. & Corvera, S. What causes the insulin resistance underlying obesity? Curr. Opin. Endocrinol. Diabetes Obes. 19, 81–87 (2012).
Stefan, N., Häring, H.U., Hu, F.B. & Schulze, M.B. Metabolically healthy obesity: epidemiology, mechanisms, and clinical implications. Lancet Diabetes Endocrinol. 1, 152–162 (2013).
Robbins, A.L. & Savage, D.B. The genetics of lipid storage and human lipodystrophies. Trends Mol. Med. 21, 433–438 (2015).
Semple, R.K., Savage, D.B., Cochran, E.K., Gorden, P. & O'Rahilly, S. Genetic syndromes of severe insulin resistance. Endocr. Rev. 32, 498–514 (2011).
Samuel, V.T., Petersen, K.F. & Shulman, G.I. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375, 2267–2277 (2010).
Danforth, E. Jr. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat. Genet. 26, 13 (2000).
Unger, R.H. Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol. Metab. 14, 398–403 (2003).
Virtue, S. & Vidal-Puig, A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome—an allostatic perspective. Biochim. Biophys. Acta 1801, 338–349 (2010).
Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Invest. 106, 171–176 (2000).
Karpe, F. & Pinnick, K.E. Biology of upper-body and lower-body adipose tissue—link to whole-body phenotypes. Nat. Rev. Endocrinol. 11, 90–100 (2015).
Robbins, D.C. et al. The effect of diet on thermogenesis in acquired lipodystrophy. Metabolism 28, 908–916 (1979).
Shimomura, I., Hammer, R.E., Ikemoto, S., Brown, M.S. & Goldstein, J.L. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 401, 73–76 (1999).
Oral, E.A. et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 346, 570–578 (2002).
Gavrilova, O. et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J. Clin. Invest. 105, 271–278 (2000).
Kim, J.Y. et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Invest. 117, 2621–2637 (2007).
Gray, S.L. et al. Leptin deficiency unmasks the deleterious effects of impaired peroxisome proliferator-activated receptor γ function (P465L PPARγ) in mice. Diabetes 55, 2669–2677 (2006).
Medina-Gomez, G. et al. PPARγ2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 3, e64 (2007).
Knowles, J.W. et al. Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J. Clin. Invest. 126, 403 (2016).
Scott, R.A. et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 44, 991–1005 (2012).
Manning, A.K. et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 44, 659–669 (2012).
Salazar, M.R. et al. Comparison of the abilities of the plasma triglyceride/high-density lipoprotein cholesterol ratio and the metabolic syndrome to identify insulin resistance. Diab. Vasc. Dis. Res. 10, 346–352 (2013).
Scott, R.A. et al. Common genetic variants highlight the role of insulin resistance and body fat distribution in type 2 diabetes, independent of obesity. Diabetes 63, 4378–4387 (2014).
Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013).
Prokopenko, I. et al. A central role for GRB10 in regulation of islet function in man. PLoS Genet. 10, e1004235 (2014).
Morris, A.P. et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 44, 981–990 (2012).
Nikpay, M. et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130 (2015).
Shungin, D. et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015).
Kozaki, K. et al. Mutational analysis of human lipoprotein lipase by carboxy-terminal truncation. J. Lipid Res. 34, 1765–1772 (1993).
Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N. Engl. J. Med. 374, 1134–1144 (2016).
Mailly, F. et al. A common variant in the gene for lipoprotein lipase (Asp9→Asn). Functional implications and prevalence in normal and hyperlipidemic subjects. Arterioscler. Thromb. Vasc. Biol. 15, 468–478 (1995).
Avila, M. et al. Clinical reappraisal of SHORT syndrome with PIK3R1 mutations: towards recommendation for molecular testing and management. Clin. Genet. http://dx.doi.org/10.1111/cge.12688 (2015).
Thauvin-Robinet, C. et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am. J. Hum. Genet. 93, 141–149 (2013).
Chudasama, K.K. et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am. J. Hum. Genet. 93, 150–157 (2013).
Dyment, D.A. et al. Mutations in PIK3R1 cause SHORT syndrome. Am. J. Hum. Genet. 93, 158–166 (2013).
Pers, T.H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015).
Lane, J.M., Doyle, J.R., Fortin, J.P., Kopin, A.S. & Ordovás, J.M. Development of an OP9 derived cell line as a robust model to rapidly study adipocyte differentiation. PLoS One 9, e112123 (2014).
Yaghootkar, H. et al. Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes 63, 4369–4377 (2014).
Lu, Y. et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat. Commun. 7, 10495 (2016).
Kilpeläinen, T.O. et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat. Genet. 43, 753–760 (2011).
Yaghootkar, H. et al. Genetic evidence for a link between favorable adiposity and lower risk of type 2 diabetes, hypertension and heart disease. Diabetes 65, 2448–2460 (2016).
Biggs, M.L. et al. Association between adiposity in midlife and older age and risk of diabetes in older adults. J. Am. Med. Assoc. 303, 2504–2512 (2010).
Pischon, T. et al. General and abdominal adiposity and risk of death in Europe. N. Engl. J. Med. 359, 2105–2120 (2008).
Vague, J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am. J. Clin. Nutr. 4, 20–34 (1956).
Smith, U. Abdominal obesity: a marker of ectopic fat accumulation. J. Clin. Invest. 125, 1790–1792 (2015).
Després, J.P. & Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 444, 881–887 (2006).
Dahlman, I. et al. Numerous genes in loci associated with body fat distribution are linked to adipose function. Diabetes 65, 433–437 (2016).
Rydén, M., Andersson, D.P., Bergström, I.B. & Arner, P. Adipose tissue and metabolic alterations: regional differences in fat cell size and number matter, but differently: a cross-sectional study. J. Clin. Endocrinol. Metab. 99, E1870–E1876 (2014).
Pinnick, K.E. et al. Distinct developmental profile of lower-body adipose tissue defines resistance against obesity-associated metabolic complications. Diabetes 63, 3785–3797 (2014).
Baughman, B.M., Pattenden, S.G., Norris, J.L., James, L.I. & Frye, S.V. The L3MBTL3 methyl-lysine reader domain functions as a dimer. ACS Chem. Biol. 11, 722–728 (2016).
Randall, J.C. et al. Sex-stratified genome-wide association studies including 270,000 individuals show sexual dimorphism in genetic loci for anthropometric traits. PLoS Genet. 9, e1003500 (2013).
Paternoster, L. et al. Adult height variants affect birth length and growth rate in children. Hum. Mol. Genet. 20, 4069–4075 (2011).
Wood, A.R. et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186 (2014).
Gupta, G.D. et al. A dynamic protein interaction landscape of the human centrosome–cilium interface. Cell 163, 1484–1499 (2015).
Singaraja, R.R. et al. Identification of four novel genes contributing to familial elevated plasma HDL cholesterol in humans. J. Lipid Res. 55, 1693–1701 (2014).
Chasman, D.I. et al. Forty-three loci associated with plasma lipoprotein size, concentration, and cholesterol content in genome-wide analysis. PLoS Genet. 5, e1000730 (2009).
DeFronzo, R.A. et al. Pioglitazone for diabetes prevention in impaired glucose tolerance. N. Engl. J. Med. 364, 1104–1115 (2011).
DREAM (Diabetes REduction Assessment with ramipril and rosiglitazone Medication) Trial Investigators. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet 368, 1096–1105 (2006).
Lincoff, A.M., Wolski, K., Nicholls, S.J. & Nissen, S.E. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. J. Am. Med. Assoc. 298, 1180–1188 (2007).
Kernan, W.N. et al. Pioglitazone after ischemic stroke or transient ischemic attack. N. Engl. J. Med. 374, 1321–1331 (2016).
Nissen, S.E. & Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 356, 2457–2471 (2007).
Food and Drug Administration. Guidance for Industry. Diabetes Mellitus—Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes (Food and Drug Administration, 2008).
Swerdlow, D.I. et al. HMG–coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. Lancet 385, 351–361 (2015).
Dewey, F.E. et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N. Engl. J. Med. 374, 1123–1133 (2016).
Do, R. et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 518, 102–106 (2015).
Jørgensen, A.B., Frikke-Schmidt, R., Nordestgaard, B.G. & Tybjærg-Hansen, A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N. Engl. J. Med. 371, 32–41 (2014).
TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med. 371, 22–31 (2014).
Myocardial Infarction Genetics Consortium Investigators. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N. Engl. J. Med. 371, 2072–2082 (2014).
Moltke, I. et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 512, 190–193 (2014).
Day, N. et al. EPIC-Norfolk: study design and characteristics of the cohort. European Prospective Investigation of Cancer. Br. J. Cancer 80 (Suppl. 1), 95–103 (1999).
Riboli, E. et al. European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr. 5, 1113–1124 (2002).
InterAct Consortium. Design and cohort description of the InterAct Project: an examination of the interaction of genetic and lifestyle factors on the incidence of type 2 diabetes in the EPIC Study. Diabetologia 54, 2272–2282 (2011).
Collins, R. What makes UK Biobank special? Lancet 379, 1173–1174 (2012).
Lyssenko, V. et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Invest. 117, 2155–2163 (2007).
Aschard, H., Vilhjálmsson, B.J., Joshi, A.D., Price, A.L. & Kraft, P. Adjusting for heritable covariates can bias effect estimates in genome-wide association studies. Am. J. Hum. Genet. 96, 329–339 (2015).
Day, F.R., Loh, P.R., Scott, R.A., Ong, K.K. & Perry, J.R. A robust example of collider bias in a genetic association study. Am. J. Hum. Genet. 98, 392–393 (2016).
Burgess, S., Butterworth, A. & Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Lawrence, R.D. Types of human diabetes. BMJ 1, 373–375 (1951).
Herbst, K.L. et al. Köbberling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care 26, 1819–1824 (2003).
Payne, F. et al. Hypomorphism in human NSMCE2 linked to primordial dwarfism and insulin resistance. J. Clin. Invest. 124, 4028–4038 (2014).
UK10K Consortium. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Delaneau, O., Marchini, J. & Zagury, J.F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2011).
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39, 906–913 (2007).
Pruim, R.J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Ward, L.D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Johnson, A.D. et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24, 2938–2939 (2008).
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP–trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014).
Zhang, X. et al. Synthesis of 53 tissue and cell line expression QTL datasets reveals master eQTLs. BMC Genomics 15, 532 (2014).
Buil, A. et al. Gene–gene and gene–environment interactions detected by transcriptome sequence analysis in twins. Nat. Genet. 47, 88–91 (2015).
GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Nica, A.C. et al. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 6, e1000895 (2010).
Acknowledgements
We are grateful for the OP9-K cells kindly shared by the laboratory of A. Kopin (Tufts Medical Center). The authors gratefully acknowledge the help of the MRC Epidemiology Unit Support Teams, including the Field Teams, the Laboratory Team and the Data Management Team, and of the staff of the Wellcome Trust Clinical Research Facility.
This study was funded by the UK Medical Research Council through grants MC_UU_12015/1, MC_PC_13046, MC_PC_13048 and MR/L00002/1. This work was supported by the MRC Metabolic Diseases Unit (MC_UU_12012/5) and the Cambridge NIHR Biomedical Research Centre and EU/EFPIA Innovative Medicines Initiative Joint Undertaking (EMIF grant 115372). Funding for the InterAct project was provided by the EU FP6 program (grant LSHM_CT_2006_037197). This work was funded, in part, through an EFSD Rising Star award to R.A.S. supported by Novo Nordisk. D.B.S. is supported by Wellcome Trust grant 107064. M.I.M. is a Wellcome Trust Senior Investigator and is supported by the following grants from the Wellcome Trust: 090532 and 098381. M.v.d.B. is supported by a Novo Nordisk postdoctoral fellowship run in partnership with the University of Oxford. I.B. is supported by Wellcome Trust grant WT098051. S.O'R. acknowledges funding from the Wellcome Trust (Wellcome Trust Senior Investigator Award 095515/Z/11/Z and Wellcome Trust Strategic Award 100574/Z/12/Z).
AUTHOR CONTRUBUTIONS
Concept and design: L.A.L., I.B., N.J.W., D.B.S., C.L., S.O'R. and R.A.S. Generation, acquisition, analysis and/or interpretation of data: all authors. Drafting of the manuscript: L.A.L., I.B., N.J.W., D.B.S., C.L., S.O'R. and R.A.S. Critical review of the manuscript for important intellectual content and approval of the final version of the manuscript: all authors.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 2 Design of the study, investigated phenotypes, sources of data and sample size.
The reported sample size is the maximum available for a given trait or set of traits in this study. *In the study by Knowles and colleagues (PubMed ID: 25798622), insulin sensitivity was measured by euglycemic clamp or insulin suppression test in 2,764 European individuals from four cohorts. IR, insulin resistance; FIadjBMI, fasting insulin levels adjusted for body mass index; TG, triglyceride levels; HDL, high-density lipoprotein cholesterol levels; ISI, insulin sensitivity index; DEXA, dual-energy X-ray absorptiometry; BF%, body fat percentage; FPLD1, familial partial lipodystrophy type 1; MAGIC, Meta-Analyses of Glucose- and Insulin-related traits Consortium; GLGC, Global Lipids Genetics Consortium; GIANT, Genetic Investigation of ANthropometric Traits; DIAGRAM, DIAbetes Genetics Replication And Meta-analysis; CARDIOGRAM, Coronary ARtery DIsease Genome-wide Replication and Meta-analysis; C4D, Coronary Artery Disease Genetics consortium.
Supplementary Figure 3 Combined directional Manhattan plots of the association with insulin-resistance-related phenotypes.
(a–c) Manhattan plots of the association of SNPs with fasting insulin adjusted for body mass index (FIadjBMI) (a), triglycerides (b) and HDL cholesterol (c). We plotted only variants with FIadjBMI, triglycerides and HDL cholesterol (P < 0.005 for each phenotype). All associations are represented for the FIadjBMI-raising allele. The 630 alleles associated with higher FIadjBMI, higher triglycerides and lower HDL cholesterol are plotted in dark red. The graph also plots 21 variants that met the P-value threshold for the three phenotypes but were not associated in the required direction (gray). For graphic display purposes, P values below 10−20 are represented as 10−20.
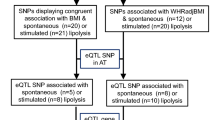
Supplementary Figure 4 Flowchart of the identification of insulin resistance loci.
Numbers refer to SNPs. FIadjBMI, fasting insulin adjusted for body mass index; HDL, high-density lipoprotein cholesterol; TG, triglycerides.
Supplementary Figure 5 Associations with insulin resistance phenotypes in an independent data set.
The figure reports associations of the genetic scores comprising the 53 or 43 SNPs with fasting insulin adjusted for body mass index, triglycerides and HDL cholesterol in up to 6,101 participants of the Fenland study who were not included in any of the discovery efforts used for identification of the 53 loci. Squares indicate the central estimate of the β coefficient; error bars represent 95% confidence intervals. N, number of participants; FIadjBMI, fasting insulin adjusted for body mass index; HDL, high-density lipoprotein; SD, standard deviation.
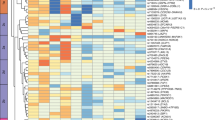
Supplementary Figure 6 Associations with glycemic and anthropometric traits and with disease endpoints at the 53 genomic loci.
The heat map represents z scores for the association of the lead insulin-raising allele at each locus. Loci are ranked on the basis of their z scores for fasting insulin (largest to smallest). With the exception of fasting insulin, none of the association analyses were adjusted for body mass index. N, maximum sample size; FIadjBMI, fasting insulin adjusted for body mass index; HDL, high-density lipoprotein cholesterol; BMI, body mass index; WHR, waist–hip ratio; CHD, coronary heart disease; T2D, type 2 diabetes. Color scale: red indicates positive associations for the insulin-raising allele at each locus, while blue indicates negative associations. Asterisks indicate known loci for the traits, i.e., those for which our lead SNP is within 500 kb on either side of a lead SNP from the largest GWAS for that trait.
Supplementary Figure 7 Associations of the genetic scores comprising the 53 or 43 SNPs with glycemic and anthropometric traits in large-scale meta-analyses and in the Fenland study.
(a) Association of the genetic scores with anthropometric and glycemic traits in meta-analyses of genetic association studies. Body mass index, waist–hip ratio, and waist and hip circumference data are from the GIANT consortium and the UK Biobank study. Body fat percentage data are from the UK Biobank, EPIC-Norfolk and Fenland studies. Fasting plasma glucose, 2-h glucose and HbA1c data are from the MAGIC consortium. Leg fat mass data are from the EPIC-Norfolk and Fenland studies. Squares with error bars represent the per-allele β coefficients in standard deviation units and their 95% confidence intervals. (b) Association with the same traits in participants of the Fenland study not included in the discovery efforts that contributed to the identification of the 53 loci. Because HbA1c has been measured only in a subset of Fenland participants, the HbA1c analysis also includes individuals from the InterAct study subcohort who did not take part in the discovery efforts that contributed to the identification of the 53 loci. Squares with error bars represent the per-allele β coefficients in standard deviation units and their 95% confidence intervals. Red and blue squares represent the results of the 53-SNP and 43-SNP genetic scores, respectively. None of the results presented in the figure were adjusted for body mass index. N, number of participants; SD, standard deviation; BMI, body mass index; WHR, waist–hip ratio; FPG, fasting plasma glucose.
Supplementary Figure 8 Associations of the 53-SNP genetic score with detailed anthropometric variables from dual-energy X-ray absorptiometry.
The figure represents the association of quintiles of the 53-SNP genetic score with the absolute values of compartmental and total fat mass. Data are from 9,747 participants of the Fenland study. The Fenland population was divided into quintiles of the distribution of the genetic score, and each quintile was compared with the bottom (reference category). Squares with error bars represent the β coefficients in grams for individuals in the exposure category as compared with the reference category and their 95% confidence intervals.
Supplementary Figure 9 Associations of the rs4976033[G] allele near PIK3R1 with continuous metabolic traits and cardiometabolic disease endpoints.
(a) Associations with continuous traits. (b) Associations with disease endpoints. Squares with error bars represent the β coefficients (a) or odds ratios (b) and their 95% confidence intervals. HDL, high-density lipoprotein; LDL, low-density lipoprotein; BMI, body mass index; WHR, waist–hip ratio; FIadjBMI, fasting insulin adjusted for BMI; FPG, fasting plasma glucose; 2hr glucose, glucose at 2 h during an oral glucose challenge; SD, standard deviation; OR, odds ratio.
Supplementary Figure 10 Associations of functional variants in LPL with cardiometabolic traits and disease endpoints.
(a) Association of the gain-of-function p.Ser447* (rs328; left) and the loss-of-function p.Asp36Asn (rs1801177; right) variants in LPL with lipid levels, anthropometric traits, liver markers and glycemic traits. (b) Association of the two variants with the risk of coronary heart disease (from the Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators; PubMed ID: 26934567) and that of type 2 diabetes. Squares with error bars represent the β coefficients (a) or odds ratios (b) and their 95% confidence intervals. HDL, high-density lipoprotein; LDL, low-density lipoprotein; BMI, body mass index; WHR, waist–hip ratio; VAT, visceral adipose tissue; ALT, alanine aminotransferase; GGT, γ-glutamyltransferase; FIadjBMI, fasting insulin adjusted for BMI; FPG, fasting plasma glucose; 2hr, glucose at 2 h during an oral glucose challenge; SD, standard deviation; OR, odds ratio.
Supplementary Figure 11 Mechanistic hypothesis for the implication of putative effector genes in the observed associations and selection of genes for experimental validation in cellular models of adipogenesis.
(a) Mechanistic hypothesis. (b) Selection criteria to prioritize genes for experimental validation. (c) Selection flowchart. Numbers in c refer to loci meeting certain selection criteria. *We did not take forward the KLF14 gene to experimental validation because previous studies on the role of this gene in metabolic disease suggest complex etiological mechanisms at this locus, including a possible parent-of-origin effect.
Supplementary Figure 12 Associations with fasting insulin adjusted for body mass index, body mass index or fasting insulin of the 53 polymorphisms identified in this study.
(a) Association of the 53 lead polymorphisms from our study with FIadjBMI as a function of the association with BMI. There was no clear bias in the association with FIadjBMI (linear regression between the β coefficients of the 53 polymorphisms, P = 0.26). (b) Association with FI (unadjusted for BMI) of the lead 53 polymorphisms as a function of the association with FIadjBMI. The line of fit was aligned with the line of equality consistent with no bias. In a, the dark red line and surrounding areas represent the line of fit with 95% confidence areas. The dashed gray line in b represents the line of equality. Data on fasting insulin associations are from the MAGIC consortium; data on BMI associations are from the GIANT consortium.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–13, Supplementary Tables 1–3, 5–8 and 12–14, and Supplementary Note. (PDF 2691 kb)
Supplementary Tables 4 and 9–11
Supplementary Tables 4 and 9–11. (XLSX 266 kb)
Rights and permissions
About this article

Cite this article
Lotta, L., Gulati, P., Day, F. et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 49, 17–26 (2017). https://doi.org/10.1038/ng.3714
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3714
This article is cited by
-
The association between the AIP and undiagnosed diabetes in ACS patients with different body mass indexes and LDL-C levels: findings from the CCC-ACS project
Cardiovascular Diabetology (2024)
-
Connections between body composition and dysregulation of islet α- and β-cells in type 2 diabetes
Diabetology & Metabolic Syndrome (2024)
-
Defining type 2 diabetes polygenic risk scores through colocalization and network-based clustering of metabolic trait genetic associations
Genome Medicine (2024)
-
White adipocyte dysfunction and obesity-associated pathologies in humans
Nature Reviews Molecular Cell Biology (2024)
-
Mendelian randomization study on insulin resistance and risk of hypertension and cardiovascular disease
Scientific Reports (2024)
