
Abstract
Glioblastoma (GBM) is the most common and aggressive primary brain tumor. To better understand how GBM evolves, we analyzed longitudinal genomic and transcriptomic data from 114 patients. The analysis shows a highly branched evolutionary pattern in which 63% of patients experience expression-based subtype changes. The branching pattern, together with estimates of evolutionary rate, suggests that relapse-associated clones typically existed years before diagnosis. Fifteen percent of tumors present hypermutation at relapse in highly expressed genes, with a clear mutational signature. We find that 11% of recurrence tumors harbor mutations in LTBP4, which encodes a protein binding to TGF-β. Silencing LTBP4 in GBM cells leads to suppression of TGF-β activity and decreased cell proliferation. In recurrent GBM with wild-type IDH1, high LTBP4 expression is associated with worse prognosis, highlighting the TGF-β pathway as a potential therapeutic target in GBM.
Similar content being viewed by others


Main
GBM is the most common and most aggressive type of primary brain tumor in adults1. Therapeutic options are limited, consisting of surgery and treatment with radiotherapy plus an oral alkylating agent, temozolomide (TMZ). Despite the benefits from TMZ, the extension of patient survival averages ∼2.5 months and tumors invariably recur, leading to a fatal outcome2. Recent reports using large-scale sequencing approaches have characterized the genomic landscape of untreated tumors3,4, yet very few studies have analyzed recurrent GBM and patient cohorts are limited in size5,6,7.
The evolution of tumor cells under therapy can be viewed as a Darwinian process of clonal replacement8,9,10 in which treatment ablates vulnerable cells while positively selecting for resistant clones. Studies of spatially distinct tumor fragments indicate that treatment failure is frequently complicated by intratumoral heterogeneity (ITH), a common phenomenon in low- and high-grade glioma11,12,13,14. Mutations in TP53 were recently proposed as a marker of subclonal heterogeneity in GBM7, but a clear pattern of tumor evolution remains elusive. ITH and diversity in evolutionary trajectories preclude the identification of general evolutionary patterns in GBM, especially when only limited cohorts of patients are available.
To find genetic markers of progression and to elucidate the diverse evolutionary trajectories by which GBM can occur and recur, we performed whole-exome and transcriptome analyses of untreated and recurrence tumors from 114 patients with GBM for whom corresponding matched normal tissue was available.
Results
Longitudinal mutational landscape of GBM
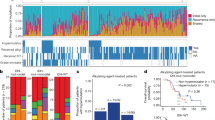
To elucidate the mechanisms driving the evolution of high-grade glioma under therapy, we analyzed 293 whole exomes and 141 transcriptomes from longitudinal tumor–matched normal samples in 114 patients with GBM (Fig. 1a). Patients with recurrent GBM (89 diagnosed with primary GBM) were collected from the Istituto Neurologico C. Besta (INCB; R001–R019), the MD Anderson Cancer Center (R020–R029), The Cancer Genome Atlas (TCGA; R030–R042), the University of California San Francisco (UCSF; R043–R052), Kyoto University (KU; R053–R055), and Samsung Medical Center (SMC; R056–R114). Whole-exome triplets comprising the initial tumor sample, recurrence tumor sample, and normal genomic DNA were sequenced for 93 patients. The transcriptomes of initial and recurrence tumor pairs were sequenced for 65 patients. All but 14 patients received standard treatment, including TMZ2. Greater than 200-fold mean target coverage was achieved in 84% of samples (246 of 293). On average, 76% of coding bases in the exome were covered by at least 100 high-quality reads (Supplementary Table 1).

(a) Number of somatic mutations. Data are shown for 114 patients from six sources (Istituto Neurologico C. Besta, MD Anderson Cancer Center, The Cancer Genome Atlas, University of California San Francisco, Kyoto University, and Samsung Medical Center). (b) Clinical and genetic profiles of patients. TMZ, treatment with temozolomide; MMR, mutation in a mismatch-repair pathway gene (MSH6, MSH2, MSH4, MSH5, PMS1, PMS2, MLH1, and MLH3 were considered); MUT, somatic nonsynonymous mutation with variant allele frequency (VAF) >5% in at least one sample; AMP and DEL, amplification or deletion with mean segmentation >0.5, as computed by SNP array data, by array comparative genomic hybridization (CGH) data or by whole-exome sequencing data. (c) Pyramid plot highlighting the correlation between different features. A hypergeometric test was performed for each pair of elements, considering initial and recurrent tumors separately. The size of each circle corresponds to the significance level of the correlation. Associations with P < 0.1 are shown. MET, altered methylation. (d) Three-dimensional bubble plot showing the frequency of somatic nonsynonymous mutations exclusively in initial tumors (red; left axis), exclusively in recurrent tumors (black; right axis), and in common to the two tumors (yellow; upper axis). Ninety-three patients with exome sequencing data from matched normal, initial tumor, and recurrent tumor samples were considered in this analysis.
To identify somatic single-nucleotide variants (SNVs) as well as short insertions and deletions (indels), we used the variant calling software SAVI2 (ref. 15). We included as somatic variants only those with a mutant allele frequency of 5% or greater. Among these variants, we selected 40 mutations from the INCB cohort for validation. Sanger sequencing successfully validated 98% (39/40) of the mutational calls as well as changes in allele frequency between untreated and recurrence tumors (Supplementary Table 2 and Supplementary Data). Untreated tumor samples harbored an average of 60 somatic mutations. Recurrence tumor samples had 585 somatic mutations on average, but this figure is not representative because of the inclusion of 17 patients (6 primary GBM and 11 secondary GBM) with hypermutated recurrence tumors (>500 mutated genes per tumor). The remaining non-hypermutated tumors had only 50 mutations on average. All hypermutated tumors originated in TMZ-treated patients. Sixteen of the 17 hypermutated samples gained mutations in genes encoding DNA mismatch-repair (MMR) proteins (MSH6, MSH2, MHS4, MSH5, PMS1, PMS2, MLH1, and MLH2) (Fig. 1b).
We compared mutations found in the initial and recurrence samples for each of the 93 patients for whom whole-exome triplets were available. Appearance of the same mutation in both the initial and recurrence samples for a patient suggests that the mutation originated relatively early in that patient's tumor development, whereas appearance of a mutation in only one sample suggests that the mutation may have occurred after the clonal lineages leading to the two samples diverged. We discovered that the mutations occurring in only one of a patient's two GBM samples outnumbered the shared mutations in more than half of all patients (57%; 53/93) (Fig. 1a, single-sample mutations versus shared mutations). We next assessed the pairwise co-occurrence and mutual exclusivity of genomic and clinical features across all patients. In addition to previously reported association16,17,18 (Fig. 1c), we observed a number of significant associations not previously reported for GBM that were exclusive to recurrence. These associations included co-occurrence of MGMT promoter methylation and hypermutation (P = 4 × 10−3, Fisher's exact test; only in TMZ-treated patients), co-deletion of RB1 and PTEN (P < 1 × 10−4, Fisher's exact test), and co-mutation of NF1 and TP53 (P = 1 × 10−2, Fisher's exact test).
Overall, our mutational analysis identifies both known and potentially new driver gene mutations in GBM. We observed mutations in known drivers of GBM3, including TP53, PTEN, EGFR, PIK3CA, ATRX, IDH1, PIK3R1, and PDGFRA, with similar frequencies in untreated and recurrence tumors (Fig. 1d). We also identified hotspot mutations in unreported potential driver genes in GBM. In particular, we found seven patients with nonsynonymous mutations in PTPN11 (encoding SHP2 protein) mapping to the first SH2 and PTP domains with a similar distribution to variants found in juvenile myelomonocytic leukemia19. A few genes seemed to be mutated and expressed exclusively in recurrence tumors (Fig. 1d), including LTBP4 (10/93), the DNA MMR gene MSH6 (encoding MutS homolog 6; 8/93), PRDM2 (10/93), and IGF1R (9/93) (for a complete list, see Supplementary Table 2). Interestingly, all eight cases with mutations in MSH6 corresponded to hypermutated recurrences (P < 1 × 10−4, Fisher's exact test), and three of these cases included nonsense mutations in the gene, indicating that loss of function of MSH6 is related to genomic hypermutation in GBM. This finding is consistent with previous observations of the induction of a hypermutated genotype following treatment of glioma20,21,22,23. The recurrence-only mutated gene LTBP4 has been reported to be an activator of transforming growth factor (TGF)-β signaling by promoting the assembly, secretion, and targeting of sites where TGF-β1 is stored and/or activated24. Disruption of Ltbp4 causes abnormal lung development, cardiomyopathy, and colorectal cancer in mice25.
To explore copy number variations (CNVs) of initial and recurrent GBM, recurrence-based analysis, GISTIC2 (ref. 26) (Supplementary Fig. 1 and Supplementary Table 3), and MutComFocal27 (Supplementary Fig. 2 and Supplementary Table 4) were applied. We found CNVs in several well-known GBM driver genes. EGFR amplification, which frequently co-occurred with EGFR SNVs and the EGFRvIII receptor variant, was observed in 42% of initial tumors (44/104) and 34% of recurrence tumors (35/102), whereas CDK4 amplification was detected in 19% of both initial and recurrence samples (20/104). Deletions in CDKN2A were the most frequent deletion in 47% of initial samples (49/104) and 52% of recurrent tumors (53/102). PTEN displayed a similar prevalence of loss in initial (37%; 38/104) and recurrent (34%; 35/102) samples.
We then defined a zygosity score to identify regions of loss of heterozygosity (LOH) (Online Methods, Supplementary Fig. 3, and Supplementary Table 5). The median zygosity score for a normal diploid chromosome is expected to be close to 0.25. To identify potential tumor suppressors associated with a two-hit mechanism, we analyzed genes with point mutations in regions with LOH in non-hypermutated recurrence tumors. This analysis recapitulated known tumor suppressors in GBM, including TP53 (14/78 samples), PTEN (9/78 samples), and NF1 (3/78 samples), and identified LOH encompassing inactivating mutations in other genes not previously reported in GBM, including APC (mutation encoding p.Arg876*) (Supplementary Table 5).
The gene fusions detected by RNA-seq analysis are summarized in Supplementary Table 6. We found gene fusions reported as recurrent alterations in GBM, such as FGFR3-TACC3 (ref. 28) and EGFR fusions with multiple partners3. FGFR3-TACC3 fusions were highly expressed in both the untreated and matched recurrence tumors, thus confirming the clonal nature of these fusion events28,29. We also found rare fusions involving other receptor tyrosine kinase (RTK)-encoding genes, such as PDGFRA, MET, and ROS. Interestingly, two patients harbored in-frame gene fusions involving MGMT at relapse (Supplementary Fig. 4). Patient R114 harbored two highly expressed in-frame fusions, NFYC-MGMT and BTRC-MGMT, and patient R056 at recurrence harbored a SAR1A-MGMT fusion. Of particular note, these three fusion transcripts carried the same breakpoint in the MGMT gene, and the reconstructed ORF for each preserved the methyltransferase and DNA-binding domains (Supplementary Table 6). The fusion transcripts were further validated by RT–PCR (Online Methods and Supplementary Fig. 5). MGMT is a gene that encodes an O6-methylguanine DNA methyltransferases, and epigenetic silencing of this gene has been associated with longer overall survival in patients with GBM under therapy30. Consistently, we observed that MGMT methylation at diagnosis predicted longer survival (P = 0.018). We also observed a high correlation between MGMT methylation and expression, both in initial and relapse samples (P = 6 × 10−3 in initial and 0.016 in relapse samples; Supplementary Fig. 6). At recurrence but not in the initial tumor, low expression of MGMT was significantly related to better prognosis (P = 4 × 10−4).
Hypermutation related to temozolomide
As shown in Figure 1a, 17% of patients with TMZ-treated GBM (17/100) relapsed with hypermutated tumors, yet there was no incidence of hypermutation in non-TMZ-treated patients (0/14). The median survival time of patients with hypermutated primary GBM having wild-type IDH1 was 24 months, corresponding to a slight increase in comparison to other patients with GBM having wild-type IDH1 (18 months). The gain of mutations in the MMR pathway as well as the accompanying hypermutation in patients with glioma after treatment has been reported before20,21,22,23, but the pattern of the hypermutated genes and the mechanism causing the mutations remain unclear. To better investigate patient mutational variation, we grouped all mutations into four types: those identified in recurrence tumors without TMZ treatment, those identified in untreated tumors, those identified in TMZ-treated but non-hypermutated cases, and those identified in TMZ-treated hypermutated cases. Hypermutated recurrence tumors were highly enriched for C>T (G>A) transitions (Fig. 2a). To identify additional markers of hypermutation, we extracted 10 bp of DNA sequence from the coding strand of hypermutated loci. Motif analysis31 showed that hypermutation occurred predominantly on the coding strand at the first cytosine of CpY elements (where Y represents a pyrimidine base) (Fig. 2b). By contrast, a pattern of mutations at CpR elements (where R represents a purine base)32 could be seen in all other tumor types tested. Although we found no significant association of the ratios of silent/missense mutations in non-hypermutated tumors, recurrence hypermutated samples contained significantly greater numbers of silent mutations (Fig. 2c). Moreover, hypermutation may be related to expression of genes involved in tumor recurrence. In hypermutated tumors, genes containing hypermutated loci were more highly expressed than mutated genes with no hypermutated loci and non-mutated genes (Fig. 2d).

(a) Fractions of the different types of nucleotide changes. In this analysis, 93 patients with data for trios of normal, initial tumor, and recurrent tumor DNA samples were considered. HM, hypermutation. (b) Hypermutation score plotted against mutation load. The logos for hypermutated and non-hypermutated samples were calculated separately on the basis of all substitutions from the respective samples. Given these logos, the hypermutation score of each sample was defined on the basis of the sample's mutation pattern. If mutations in a sample follow the pattern of the hypermutation logo, the sample will have a higher hypermutation score (in this analysis, 15 samples are in the hypermutated category). (c) Silent/missense ratio analysis. P values were calculated by rank-sum test. Patients with fewer than ten mutations in either the initial or recurrent sample were not considered for the analyses in b and c. (d) Comparison of expression levels among three gene clusters: hypermutated (HM) genes, mutated (M) genes, and non-mutated (NM) genes. The mean expression levels of the three gene clusters in samples with expression data available (m = 160) were calculated to generate the box plot. The bottom and top of each box correspond to the first and third quartiles, and the line inside is the median. Whiskers correspond to 1.5 times the IQR. P values were calculated by rank-sum test.
Reconstruction of the main routes of GBM evolution
The number of mutations exclusive to untreated tumors and recurrence tumors or in common can be used to describe an evolutionary tree. We developed a method to perform statistics on the space of evolutionary trees33 by embedding each tree in a sphere (Fig. 3a). In each representation, the upper vertex corresponds to the fraction of mutations that are common to both samples, the left vertex corresponds to the fraction of mutations exclusive to the untreated sample, and the right vertex corresponds to the fraction of mutations exclusive to recurrence. Unsupervised clustering of the different phylogenies identified three clusters (Online Methods). The yellow cluster represents the limiting case where few mutations are lost from tumors after diagnosis, similar to the classical model of linear tumor evolution typified in previous studies of treatment-naive colon cancer34. However, treatment can change linear patterns of evolution35. The abundance of points far from the right edge of the diagram suggests that, in most patients, the dominant clones before treatment are replaced by new clones that do not have many of the same mutations.

(a) Moduli space of GBM evolutionary trees. Each ball represents one patient, and the different colors correspond to three clusters in moduli space. (b) Model of branching tumor evolution. This model assumes an independent monophyletic origin for initial and recurrence tumors sharing an ancestral clonal lineage (duration tS), after which point they branch off from one another (durations tI and tR1 + tR2). After this clonal evolution, the lineage leading to each sample diversifies for duration tMRCA, during which subclonal variants can accrue. Somatic variants accrue according to the substitution rates μ1 and μ2 before and after treatment, respectively. (c) Relationship between estimated substitution rates before and after treatment (the median and IQR are shown for each patient). The dashed line is the diagonal (where the pretreatment and post-treatment substitution rates are equal). Red, hypermutated tumors; light blue, non-hypermutated tumors; squares, primary GBM diagnoses; diamonds, secondary GBM diagnoses. A black dot in the center of a symbol indicates patients whose disease fit the model well. A yellow halo indicates patients with TP53 mutated in both the initial and recurrence samples. Patient R069 was not considered for evolutionary analysis as no valid mutations were detected in the initial sample. (d) Cross-sectional integration of longitudinal data by TEDG. Arrows show the order in which mutations occur. Thicker arrows correspond to more independent patients with the same order of mutations. The size of each node corresponds to the frequency of mutations in that gene in our cohort.
If many mutations in the initial sample are lost at recurrence, this suggests that the dominant clone at recurrence originated (that is, diverged from the dominant clone at diagnosis) at a time point relatively long before the initial sample was taken. Consistent with epidemiological observations and classical models of tumor evolution from Armitage and Doll10 and Nordling9, the number of mutations in the untreated tumor increased with the patient's age at diagnosis (with an increase on average of 0.6 protein-changing mutations per year, or 0.02 mutations per megabase per year; Supplementary Fig. 7d). Encouraged by this concordance, we developed a mathematical model of branching tumor evolution with an independent monophyletic origin for diagnostic and relapse samples.
In our branching model (Fig. 3b), if a mutation occurs along the lineage common to both the initial and recurrence samples, it will be clonal in both of these samples. If a mutation occurs along the lineage leading to the most recent common ancestor (MRCA) of a single sample, then it will be clonal in that sample and absent in the other. If a mutation occurs in a descendant of the MRCA of a sample, then it will be subclonal in that sample and absent in the other. Any other pattern—where a mutation appears subclonally in both samples or appears clonally in one sample and subclonally in the other—would require either recurrent mutation or 'back-mutation' (a mutation that restores the wild-type allele). An alternate model is also possible in which the recurrence sample stems from a lineage nested within the initial sample, perhaps selected by therapy. In this case, a single mutational event could produce a variant that is present subclonally at diagnosis and clonally at recurrence, but two events would be needed to explain loss of a clonal mutation (Supplementary Fig. 7a,b). We found that 59% of patients (54/92) had at least four clonal mutations at diagnosis that were lost in the recurrence sample, supporting the branching model as the typical scenario. The picture is nuanced, however, as 17 patients had at least four subclonal mutations at diagnosis that became clonal at recurrence, supporting the alternate model as a minority scenario (Supplementary Fig. 7c).
By accounting for the likelihood of each mutational pattern in our branching model, we fit substitution rates before and after treatment, as well as the amount of time before diagnosis that the untreated and recurrence lineages diverged (Online Methods). Using a collection of statistical criteria (Online Methods), we found that 49% of the patients analyzed (45/92) fit the model well, without requiring an unrealistic frequency of recurrent mutation or back-mutation. The pretreatment substitution rate was consistent among these 45 patients (Fig. 3c), corresponding to a median (interquartile range, IQR) of 0.028 (0.018–0.041) substitutions per megabase per year. The statistics for all 92 patients were similar, with a median (IQR) of 0.024 (0.018–0.035) substitutions per megabase per year. No relationship was observed between the substitution rate and age at diagnosis (Supplementary Fig. 8). Considerably more variation was observed in post-treatment substitution rates, with 15 of the 92 patients exhibiting remarkably higher mutation rates after treatment (Supplementary Fig. 9). All but one of these patients showed hypermutation, with over 500 variants found in the recurrence sample.
Estimates of divergence time suggested that recurrence clones diverged from untreated clones many years before disease was detected (Supplementary Fig. 7e). The median divergence time among the 45 patients whose disease fit the model well was 12.6 years (range of 2.3–50.5 years, IQR of 7.2–22.6 years). Because the remaining 47 patients may fail the model's assumption that untreated and recurrence tumors are evolutionarily distinct (that is, monophyletic), we caution that divergence time for these patients should be interpreted only as a heuristic measure of the genetic difference between a given pair of tumor samples. In general, the uncertainty in divergence time estimates was large, with the median for patients whose disease fit the model well showing a 95% confidence interval of 24 years. Still, even the lower bound of the 95% confidence interval exceeded 3 years for a majority of the patients.
To analyze the potential evolutionary trajectories of GBM under therapy, a tumor evolutionary directed graph (TEDG)36 was constructed for the 93 triplet samples. As this analysis uses as input the fraction of cells harboring a particular mutation, we estimated the purity of each tumor using ABSOLUTE37 (Supplementary Table 7) and PyClone38 (Supplementary Table 8). The resulting TEDG indicates that mutations in IDH1, PIK3CA, and ATRX are early events, mutations in TP53, NF1, and PTEN occur later, and mutations in MSH6 and LTBP4 are relapse-specific events (Fig. 3d). A more complex set of possible evolutionary trajectories appears when copy number information is included in the analysis (Supplementary Fig. 10).
Clonal replacement events are frequent in GBM
To discover the pattern of alterations in recurrent GBM as compared with untreated tumors, we performed in-depth investigations into any gains or losses of genetic alterations. The EGFR gene (encoding epidermal growth factor receptor) is known to be frequently amplified, mutated, and rearranged in untreated gliomas39. To uncover the role of EGFR alterations in GBM evolution, we applied PRADA to detect EGFR structure variance from RNA-seq data40. In examining junction reads, we found at least one junction read corresponding to EGFRvIII in 18% (12/67) of initial tumors and 11% (8/76) of recurrence tumors (Supplementary Table 9). Interestingly, nine patients lost EGFRvIII and one patient gained EGFRvIII at relapse (transcribed allelic fraction >5%), indicating, first, that the variation resulting in EGFRvIII is a late event that occurred after the clonal lineages leading to the two samples diverged and, second, that EGFRvIII is more common in initial tumors and is lost during treatment (Fig. 1d). However, EGFRvIII also occurs in recurrence. An example is provided by patient R005, whose untreated tumor harbored EGFR amplification and a mutation encoding p.Ser645Cys. The EGFR mutation encoding p.Ser645Cys was lost in the recurrence tumor and replaced by a variation resulting in EGFRvIII (Supplementary Fig. 11).
A switch between differentially mutated versions of the same gene also occurred for PDGFRA (encoding platelet-derived growth factor receptor α polypeptide), another RTK-encoding gene frequently mutated in GBM (Fig. 4b and Supplementary Fig. 11). Mutation encoding p.Glu229Lys, which is relatively common in cross-sectional mutation databases (for example, TCGA41), seemed to be a relatively late event, as it was exclusive to the recurrence sample and replaced the initial mutation encoding p.Pro443Leu (Fig. 4b). Mutational replacement also occurred in the tumor suppressor TP53 (p.Gly105Arg to p.Arg337Cys in patient R038; Fig. 4c) and in EGFR (p.Ala1201Thr to p.Gly598Val in patient R065; Fig. 4d). In total, we found that 11% (10/93) of patients with recurrent GBM had clonal replacements in key driver genes (Supplementary Fig. 12). These clonal switching events in the same gene occurred preferentially in genes known to have a role in GBM (P < 1 × 10−4; Fig. 4a). The strong association between the switching of alterations and key driver genes (EGFR, TP53, and PDGFRA) suggests (i) that some of these genes contribute to a late expansion, both in treated and untreated tumors, and (ii) that converging evolution is associated with these genes.

(a) Mutations of seven key GBM drivers (EGFR, TP53, PDGFRA, PTEN, ATRX, NF1, and RB1) were replaced by different mutations in the same genes (enrichment compared to non-drivers at P < 1 × 10−4, Fisher's exact test). (b–d) Replacement in three different patients of mutations in PDGFRA (b), TP53 (c), and EGFR (d). Cancer cell frequency was estimated by PyClone.
Expression analysis and subtype switching
On the basis of gene expression pattern, GBM is commonly divided into four subtypes, which show different responses to treatment4. To study the evolution of gene expression in GBM, we followed the ssGSEA method4 to subtype each tumor sample (Fig. 5a). As expected, we found that patients with IDH1 mutations were mostly classified as proneural gliomas42; EGFR alterations were associated with the classical subtype; and NF1 alterations were associated with the mesenchymal subtype (Fig. 5b). We observed that all five hypermutated primary GBM cases switched subtype (two to the mesenchymal, one to the neural, and two to the proneural subtypes). Strikingly, we found that two-thirds of primary GBM cases (39/58) switched transcriptional subtype at relapse, whereas secondary GBM cases seemed more stable (2/7 switched subtype) (Fig. 5a). Interestingly, the mesenchymal subtype was the most stable primary GBM subtype, switching in 55% (12/22) primary GBM cases at recurrence, and the mesenchymal subtype at recurrence was associated with worse overall survival (P = 3 × 10−3; Supplementary Fig. 13). As EGFRvIII is associated with the classical subtype (Fig. 5b), loss of this alteration in the recurrence tumor was consistently associated with transition from the classical subtype to other expression subtypes (P = 8 × 10−3, Fisher's exact test; Fig. 5a,c).

(a) Expression-based GBM subtyping. ssGSEA was performed to cluster each sample into one of four subtypes (proneural, neural, classical, and mesenchymal). An asterisk indicates samples where the subtype had a maximal enrichment score (max ES). If the optimal subtypes in the initial and recurrence tumors were different, a patient is labeled as having switched subtype. The P value was calculated by Fisher's exact test. (b) Association between switching of expression-based subtype and genetic and clinical features. The analysis was performed as in Figure 1c. (c) Stochastic matrix of GBM subtypes. The large cohort of longitudinally collected GBM samples allows the construction of a probability matrix for transitions between the four subtypes. Arrows show the frequencies at which patients stay in a subtype or switch from one subtype to another. Bottom, a stationary distribution was generated on the basis of the stochastic matrix, indicating the proportion of the four subtypes after treatment.
LTBP4 promotes tumor growth and reduces survival
We found that the LTBP4 gene (encoding latent TGF-β–binding protein 4) harbored significantly more mutations in recurrent than untreated GBM (Fig. 1d and Supplementary Fig. 14). The LTBP4 gene encodes a protein that belongs to the LTBP family, which is implicated in regulation of the TGF-β pathway, typically acting as an activator of TGF-β signaling24. Interestingly, activation of TGF-β is known to drive aggressiveness in malignant glioma43,44,45,46. We found that high expression of LTBP4 in recurrence tumors was associated with worse prognosis in patients with primary GBM having wild-type IDH1 (P = 7 × 10−3; Fig. 6b). Furthermore, mutations in LTBP4 were correlated with higher expression of this gene (P < 0.05; Fig. 6a). Further strengthening the case that LTBP4 expression could drive tumor growth via TGF-β activation, elevated expression of LTBP4 in GBM was associated with elevated expression of genes implicated in the TGF-β pathway (Gene Set Enrichment Analysis, false discovery rate (FDR) < 0.05; Fig. 6c).

(a) LTBP4 mutation is related to high expression of this gene. The P value was calculated by rank-sum test. (b) Analysis of the effect of LTBP4 expression on the survival of patients with primary GBM having wild-type IDH1. 'High' indicates a z score for LTBP4 expression at relapse of >0, whereas 'low' indicates a z score for LTBP4 expression of <0. The P value was calculated by log-rank test. Only patents with primary GBM having wild-type IDH1 were considered in this analysis. (c) Gene set enrichment analysis. Recurrence tumor samples of primary GBM with wild-type IDH1 were grouped according to LTBP4 expression. Samples with high LTBP4 expression (z score >0) were enriched for TGF-β activity. NES, normalized enrichment score. (d) Immunoblot of U87 and U251 glioma cells transduced with three independent shRNAs, two against LTBP4 (LTBP4-sh1 and LTBP4-sh2) and one non-targeting shRNA (sh-NT) as control. β-actin was used as loading control. (e,f) qRT–PCR of TGF-β target genes in U87 (e) and U251 (f) cells; n = 9 (three biological replicates performed in triplicate). Data are shown as means ± s.d.: P < 0.05. AU, arbitrary units. (g,h) Growth curves in a representative experiment using U87 (g) and U251 (h) glioma cells treated as in d. Data are shown as the means of six experimental replicates ± s.d.
To experimentally test the functional link between LTBP4 and TGF-β, we used lentiviruses carrying two independent short hairpin RNA (shRNA) cassettes targeting LTBP4 to silence the LTBP4 gene in the human glioma cell lines U87 and U251 (Fig. 6d). LTBP4 silencing in both cell lines resulted in reduced expression of the ID genes ID1 and ID2, whose expression is positively regulated by TGF-β in glioma. Conversely, LTBP4 silencing also led to the upregulation of RHOB and GADD45A, two genes repressed by TGF-β in glioma (Fig. 6e,f)43. Consistent with the protumorigenic role of TGF-β in GBM, LTBP4 silencing markedly impaired the proliferation of U87 and U251 glioma cells (Fig. 6g,h).
Discussion
Using longitudinal genomic and transcriptomic analyses of 114 patients with GBM, we have detailed the major routes of GBM evolution under therapy. GBM evolution is highly branched, and specific alterations and evolutionary patterns are associated with treatment. Our first observation from this analysis is that, despite 45% of mutations (in non-hypermutated tumors) being shared by diagnostic and relapse samples, the dominant clone at diagnosis is generally not a lineal ancestor of the dominant clone at relapse. Instead, these two clones seem to have diverged from a common ancestor more than a decade before diagnosis in most patients (Supplementary Fig. 7e).
Because 11% of patients (10/93) exhibit replacement of one mutated version of a gene (at diagnosis) with another, differently mutated version of the same gene (at relapse), it is conceivable that the genes associated with undergoing clonal completion are the sites of late driver events. In fact, this phenomenon of mutational switching is enriched ∼200-fold in genes known to be implicated in GBM, including EGFR, TP53, and PDGFRA (Fig. 4a and Supplementary Fig. 13). This scenario of convergent evolution suggests that the common ancestor of the diagnostic and relapse clones had fewer driver alterations and likely a less aggressive phenotype. The accumulation of alterations in GBM cells therefore seems to occur over a decade(s)-long growth phase that leads to a highly diverse population, with each clone experiencing a parallel series of expansions.
Related to mutational switching, we also find that two-thirds of patients with primary GBM exhibit different transcriptional subtypes at diagnosis and relapse. Our observation of subtype switching, considered together with recent findings that different parts of the same tumor can exhibit different GBM subtypes12,47, also calls into question the usefulness of the expression-based classification system as a prognostic marker before relapse.
Evolutionary dynamics generally appear similar before and after treatment: our mathematical model estimates typical substitution rates of ∼0.03 substitutions per megabase per year during both periods, except in the 16% of cases that recur with hypermutated tumors. Hypermutated tumors, which are highly enriched for mutations at CpC dinucleotides21, harbor mutations in MMR pathway genes, most commonly in MSH6, and can exhibit 100-fold higher substitution rates (∼3 substitutions per megabase per year). We found that hypermutation preferentially targets highly expressed genes, suggesting that the mutagenic mechanisms related to TMZ treatment and subsequent MMR alteration act more efficiently in highly expressed regions of open chromatin.
Finally, and of particular relevance to the discovery of new GBM treatment strategies, we uncovered unique alterations associated with relapsed GBM. In addition to identifying previously reported mutations in MMR pathway genes in 15% of patients (14/93), we found mutations in the LTBP4 gene in 11% of relapse tumors (10/93). LTBP4 encodes a protein that binds to TGF-β. The TGF-β signaling pathway has been associated with a variety of biological contexts, including cell proliferation, epithelial-to-mesenchymal transition21,48, and apoptosis. We have provided both clinical and in vitro evidence that LTBP4 seems to activate this signaling pathway to drive tumor growth: higher expression of LTBP4 in primary GBM samples with wild-type IDH1 is associated with poorer survival (P = 7 × 10−3; Fig. 6b), and silencing LTBP4 in two different cell lines decreases both cell proliferation and the expression of TGF-β target genes. These results are consistent with recent animal studies showing that TGF-β inhibitors reduce the viability and invasiveness of gliomas49 and advance the case for these molecules as potential antitumor therapeutics.
In conclusion, our study sketches the main routes of GBM evolution under therapy, identifying a highly branched process with specific alterations and evolutionary patterns associated with treated tumors.
Methods
Patients and samples.
Patients with recurrent GBM were collected from INCB (R001–R019), the MD Anderson Cancer Center (R020–R029; ref. 7), TCGA (R030–R042; ref. 7), UCSF (R043–R052; ref. 13), KU (R053–R055; ref. 14), and SMC (R056–R093 (ref. 6) and R094–R114).
The specimens in the INCB cohort originate from the Besta Brain Tumor Biobank, which is partly funded by the Italian Ministry of Health. All patients signed informed consent for the use of their biological material for research purposes. One case (R012) in this cohort had a history of lower-grade glioma before first diagnosis with GBM. All patients were treated by standard Stupp treatment with surgery followed by radiotherapy plus concomitant and adjuvant TMZ2.
Samples from the MD Anderson cohort were primary and recurrent paired tumors obtained from the Henry Ford Hospital in accordance with institutional policies, and all patients provided written consent, with approval from the Institutional Review Board (protocol 402). Three cases had a history of lower-grade astrocytoma before the first diagnosis with GBM (R022, R027, and R029). All of the recurrent GBMs had been treated with radiochemotherapy plus TMZ. The TCGA cohort contains TCGA samples, collected following the published protocol for TCGA. All of the recurrent GBMs had been treated with chemotherapy or radiation. Six patients were not treated with TMZ (R031 and R034–R038). The MD Anderson and TCGA cohorts were initially published by Kim et al.7.
The UCSF cohort contains eight patients (R043–R050) collected from the Neurosurgery Tissue Bank at UCSF, with approval by the Committee on Human Research at UCSF. Two patients (R051 and R052) in this cohort were from the University of Tokyo hospital, where the study was approved by the ethics committee of the University of Tokyo. Initial tumors from all patients in this cohort were low-grade gliomas, and their recurrences were secondary GBM. This cohort was initially published by Johnson et al.13. The KU cohort makes use of data generated by the Department of Pathology and Tumor Biology at Kyoto University. Initial tumors of patients in the KU cohort were low-grade gliomas, and their recurrences were secondary GBM. These patents were initially described by Suzuki et al.14.
The SMC cohort consists of GBM samples from the Samsung Medical Center, Republic of Korea, including samples from a previous publication (R056–R093, including 14 cases from Seoul National University Hospital; Kim et al.6) and additional, unpublished samples (R094–R114). All samples in the SMC cohort had been collected with approval from the institutional review board (files 201004004 and 201310072). Initial tumors from R076–R078, R098, R105, and R114 were secondary GBM, with a history of low-grade glioma. Patient R103 had cervical cancer 3 years before first diagnosis with GBM.
Detailed clinical information for all cohorts is provided in Supplementary Table 10.
Sequencing and mapping.
Genomic DNA from initial tumor–recurrence tumor–matched normal blood triplets for patients R001–R016 and recurrent tumors for patients R017–R019 was extracted, purified, quantified, fragmented, subjected to quality control, and used to create a library of genomic DNA fragments. Genomic DNA fragmentation was performed using the Covaris S220 AFA instrument to reproducibly generate fragments of a precise length; quality control of both genomic DNA samples and library fragments (at a later time) was performed using the Agilent Bioanalyzer 2100 microfluidic device. Both untreated and treated tumor samples from R009, R011, and R014, plus recurrent samples from patients R017–R019 were sequenced using the Agilent v3 50Mb kit, obtaining 90-bp paired-end reads. Mapping files for untreated, normal samples from patients R017–R019 were obtained from TCGA through the Cancer Genomics Hub. All other DNA samples from the INCB cohort were sequenced using the protocol for the Agilent SureSelect XT Human All Exon v4 Kit, with paired-end sequencing of 80 million reads with 150× on-target coverage. High-quality reads for these samples were mapped by BWA50 to the hg19 (ref. 51) human genome assembly with default parameters. All mapped reads were then marked for duplicates by Picard to eliminate potential duplications. Total RNA for the samples in the INCB cohort was collected to investigate transcriptional profiles by mRNA-seq using Illumina technology. After quantification and quality controls, mRNA was reverse transcribed to cDNA and a library of fragments was synthesized using Illumina TruSeq mRNA kits. Total RNA depleted of rRNA from patients R001–R005, R007, R008, R010, and R012 was sequenced by TrueSeq3 stranded prep (Illumina). RNA samples from R006, R009, and R017–R019 were sequenced at BGI. All reads were mapped to the hg19 human genome assembly from the UCSC Genome Browser51, using a fast splice junction mapper, TopHat52.
Mapping files for the TCGA samples except for R039 were downloaded through the Cancer Genomics Hub from TCGA. DNA mapping files for the UCSF, MD Anderson, and KU cohorts and patients R056–R093 from the SMC cohort were all downloaded from the European Genome-phenome Archive. (EGAS00001000579, EGAD00001001113, EGAD00001001213, and EGAD00001001424) Additional samples (R094–R114) for the SMC cohort were subjected to the same sequencing protocols as the samples described in the previous publication (R056–R093; ref. 6).
SAVI2 and driver gene selection.
To identify somatic mutations from whole-exome sequencing data for triplet samples (normal, initial tumor, and recurrence tumor) from patients with GBM, we applied the variance calling software SAVI2 (statistical algorithm for variant frequency identification15), which is based on the empirical Bayesian method. Specifically, we first generated a list of candidate variants by successively eliminating positions without variant reads, positions with low sequencing depth, positions that were biased for one strand, and positions that contained only low-quality reads. Then, the number of high-quality reads for forward-strand reference alleles, reverse-strand reference alleles, forward-strand non-reference alleles, and reverse-stand non-reference alleles was calculated at the remaining candidate positions to build the prior and the posterior distribution of mutation allele fraction. Finally, somatic mutations were identified on the basis of the posterior distribution of differences in mutation allele fraction between normal and tumor samples15. SAVI2 was able to assess mutations by simultaneously considering multiple tumor samples, as well as their corresponding RNA samples, if available.
The identity of common somatic mutations for patients R078–R082 was unknown because of the lack of normal DNA for these patients. Mutations exclusive to initial and recurrence samples were identified on the basis of differences between initial and recurrence tumor DNA. Tumor DNA from patients R083–R093, R102, and R111–R114 was not complete. The somatic mutations for these patients were estimated on the basis of RNA-seq data.
The list of known drivers used in this manuscript (Supplementary Table 2) was generated by combining GBM drivers from the Cancer Gene Census53 and our previous analysis of primary GBM2.
Analysis of loss of heterozygosity and copy number change.
All common dbSNP variants from single samples were extracted to define the zygosity score (ZS) as ZS = f(1 − f), where f is mutation allele frequency. The LOH rate (r) of somatic mutations in a tumor sample was then defined by

where  is the zygosity score in tumor samples and
is the zygosity score in tumor samples and  is the zygosity score in normal samples. If r < 0.8, we expected the corresponding mutation to be in an LOH region. Segmentation in Supplementary Figure 3 was performed with the CBS algorithm54.
is the zygosity score in normal samples. If r < 0.8, we expected the corresponding mutation to be in an LOH region. Segmentation in Supplementary Figure 3 was performed with the CBS algorithm54.
The EXCAVATOR55 pipeline was carried out to detect copy number alterations on the basis of whole-exome sequencing data. EXCAVATOR considers the mean number of reads per exon and normalizes the data by a three-step normalization procedure to eliminate bias introduced by GC content, genomic mappability, and exon size. Segmentation was then performed with a new heterogeneous hidden Markov model algorithm, the heterogeneous shifting-level model (HSLM) algorithm, which considers the genomic distance between consecutive exons55. To confidently quantify variation arising in whole-exome sequencing data in each patient's initial and recurrence samples in comparison to normal samples, we calibrated whole-exome sequencing CNV calls to SNP array data in samples for which these data were available (Supplementary Fig. 15). In addition to whole-exome sequencing, we used segmentation data for TCGA samples from the Broad Firehose platform and, when available, SNP6 data preprocessed with AROMA and normalized array CGH data obtained from the Gene Expression Omnibus (GSE63035). To identify statistically significant regions of variation, GISTIC26 was applied separately in initial and recurrence tumors. GISTIC estimated the background rates for each amplification and deletion and then summarized the input samples to score the significance of regions with altered copy number. To integrate mutation and copy number data, MutComFocal27 was separately performed in initial and recurrence tumors. In the MutComFocal analysis, long proteins (with more than 3,500 amino acids), genes lacking expression (where mutations were not expressed in any samples), and high-synonymous-rate genes (synonymous/nonsynonymous ratio >0.2) were not considered.
Gene fusion detection and structural rearrangement of EGFR.
ChimeraScan56 was used to generate the starting set of gene fusion candidates. To reduce the false positive rate and nominate potential driver events, we applied the Pegasus annotation and prediction pipeline. We reconstructed the entire fusion sequence on the basis of breakpoint coordinates and assigned a driver score to each candidate fusion via a machine learning model trained largely on GBM data57. All candidates reported in Supplementary Table 6 were selected according to three criteria: (i) Pegasus score >0.5; (ii) either more than 400 spanning reads or at least 2 split reads supporting fusion; and (iii) the two fusion partners being separated from each other by at least 50 kb.
To examine rearrangement of EGFR, we applied prada-guess-if from the PRADA package. PRADA is an RNA-seq analysis pipeline developed at MD Anderson40. Following the definition in Brennan et al., the transcribed allelic fraction of EGFRvIII was defined as the fraction of junction reads joining exon 1 to exon 8.
Gene expression analysis and expression-based subtyping analysis of GBM samples.
FPKM values were calculated by Cufflinks58. To eliminate batch effects, we normalized gene expression by calculating the z score for each batch. Gene expression was assessed by corresponding z scores for genes. ssGSEA was applied to determine the subtype of GBM samples. For each sample, z scores were used to rank all genes to generate the rnk.file as the input for the GseaPreranked software. An enrichment score was generated for all four subtypes initially defined in Verhaak et al.42. The subtype with the maximal enrichment score was selected as the representative subtype for each sample.
Moduli space analysis.
Clustering analysis of patient data was performed as follows. Each phylogenetic tree was represented as a point in the projective evolutionary moduli space, which in this case was a triple (x1, x2, x3) such that x1 + x2 + x3 = 1, by taking the raw mutation counts (z1, z2, z3) for the common, initial, and recurrent mutations and normalizing, setting xi =zi /(z1 + z2 + z3). We discarded samples for which any of the mutation counts was missing, leaving 93 points (from 114 patients). The metric for the evolutionary moduli space was in this case simply the standard Euclidean metric. Note that, for the purposes of constructing this space, the 'branch lengths' of each patient's tree were simply mutation counts, in contrast to the evolutionary analysis described below, which estimates branch lengths in years.
We then applied three clustering algorithms to this metric space: k-means clustering, spectral clustering, and density-based spatial clustering (DBSCAN). We used the code provided as part of the scikit Python package. For k-means clustering and spectral clustering, we set the number of clusters at three; DBSCAN determines the number of clusters from the data, but we set the parameters to be small distance of close neighbors ɛ = 0.5 and minimum cluster size = 5. For spectral clustering, the affinity matrix was computed using the Gaussian kernel applied to the Euclidean distance.
To ensure stability of the results, we performed cross-validation using Monte Carlo simulations in which we sampled without replacement 95% of the data points and performed clustering.
Tumor purity estimation and cellular fraction.
ABSOLUTE37 was used to infer tumor purity and ploidy for each whole-exome sequencing sample by integrating mutational allele frequencies and copy number calls.
PyClone38 was run for each sample using default parameters. Briefly, we integrated mutation alleles, copy number calls, and LOH status for each sample as input to obtain cellular frequencies. Cellular frequencies were then rescaled by median adjustment and used as input for TEDGs and mathematical modeling of tumor evolution.
Evolutionary model.
We considered all 92 patients for whom mutations were identified in both the initial and recurrence tumor samples. To exclude false positives, only variants with an allele frequency of at least 5% were used. Variants occurring at a cellular fraction of at least 95% were classified as clonal in a sample, and other variants were considered to be subclonal. Details of the model are given in the Supplementary Note. In Supplementary Figure 16, we perform sensitivity analysis using alternate cutoffs for clonality. Related code is provided as Supplementary Code.
Tumor evolutionary directed graph reconstruction.
To reconstruct the order of events during tumor progression, we followed the strategy in Wang et al.36. We selected genes that were recurrently mutated and expressed in our samples. In hypermutated cases, we only considered mutations of MSH6 and LTBP4. A mutation that was predicted to be clonal (cellular fraction >0.8) in both the initial tumor and recurrence tumor was defined as an early event, whereas a mutation that was only present (variant allele fraction >5%) in one of these samples was defined as a late event. To represent the order of clonal mutations, for each sample, directed edges were added to connect early and late events. Then, we combined the directed edges from different patients to show a global landscape of GBM evolution. A copy number alteration was defined as clonal if the absolute value of segmean was greater than 1. A copy number alteration was defined as present if above the threshold of 0.5 and as absent if below the threshold of 0.1.
Hypermutation score.
Hypermutation score (HS) was defined as

where WM is the weight matrix of the DNA sequence logo of a given sample, WMH is the weight matrix of all mutations in hypermutated samples, and WMN is the weight matrix of mutations from all non-hypermutated samples.
Validation of mutations.
The genomic regions surrounding predicted mutations were amplified using AccuPrime Taq DNA Polymerase High Fidelity (Invitrogen). Primers are summarized in Supplementary Table 11.
PCR products were purified with ExoSAP-IT (Affymetrix) and subjected to Sanger sequencing (Macrogen). The amplicons containing predicted genomic mutations were sequenced using BigDye Terminator Cycle Sequencing Kit v3.1 on the ABI Prism 3730xl DNA Analyzer (Applied Biosystems). All figures for Sanger sequencing validation are in the Supplementary Data.
To assess the sensitivity in judging absence of a mutation in one phase that is present in the other phase, we studied 15 variants in the panel that were absent in one of the samples in whole-exome sequencing, with median whole-exome sequencing depth of 117 (10–402) reads. Using CancerScan6, we found that no read reported the variant in the sample where it was deemed absent by whole-exome sequencing (Supplementary Table 12), with median CancerScan depth of 563 (217–1,377).
Cell culture, lentivirus production, and cell growth analysis.
The U87 cell line was acquired through the American Type Culture Collection (ATCC HTB-14). The U251 cell line was obtained through Sigma (09063001). Cell lines were cultured in DMEM supplemented with 10% FBS (Sigma). Cells were routinely tested for mycoplasma contamination using the Mycoplasma Plus PCR Primer Set (Agilent Technologies) and were found to be negative.
Lentivirus was generated by cotransfection of the lentiviral vectors with pCMV-ΔR8.91 and pMD2.G plasmids into HEK293T cells as previously described59,60. The sequences for the shRNAs targeting LTBP4 are provided in Supplementary Table 11.
After infection, cells were selected with puromycin (Sigma) at a concentration of 2 mg/ml for 48 h. Cells were analyzed by immunoblot analysis, qRT–PCR, and growth assay 3 d later.
Evaluation of cell growth was performed using the MTT assay. Cells were plated at a density of 2.5 × 103 cells/well in 96-well plates in six replicates and allowed to adhere for 24 h. Viability was assessed daily by adding MTT ((3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazolium, Sigma; 5 mg/ml in PBS). After 4 h of incubation, medium was removed and formazan crystals were solubilized with acidic isopropanol (0.1 N HCl in absolute isopropanol). The absorbance at 550 nm was measured with a plate reader.
RT–PCR.
Total RNA was prepared with TRIzol reagent (Invitrogen), and cDNA was synthesized using Superscript II Reverse Transcriptase (Invitrogen) as described60,61. qRT–PCR was performed with the 7500 Real-Time PCR system, using SYBR Green PCR Master Mix (Applied Biosystems). The primers used in qRT–PCR are summarized in Supplementary Table 11.
Results are presented as the means ± s.d. of three independent experiments, each performed in triplicate (n = 9). Statistical significance was determined using unequal-variance t tests (two-tailed).
Immunoblotting.
Cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1.5 mM sodium orthovanadate, 50 mM sodium fluoride, 10 mM sodium pyrophosphate, 10 mM β-glycerophosphate, and EDTA-free protease inhibitor cocktail (Roche)). Lysates were cleared by centrifugation at 25,000g for 15 min at 4 °C. Protein samples were separated by SDS–PAGE and transferred to nitrocellulose membrane. Membranes were blocked in TBS with 5% nonfat milk and 0.1% Tween-20 and probed with primary antibodies. The antibodies and working concentrations were as follows: LTBP4 (1:200 dilution; sc-393666) obtained from Santa Cruz Biotechnology; and β-actin (1:2,000 dilution; A5441) obtained from Sigma.
Gene fusion validation.
For validation of fusion transcripts, RT–PCR assays were performed. Total RNA was extracted from the tissues by AllPrep DNA/RNA Mini kit according to the manufacturer's instructions (Qiagen). Total RNA (0.5 μg) was reverse transcribed to synthesize template cDNA using a random hexamers with the Superscript III First-Strand System (Life Technologies), and 20 μl of synthesized cDNA was diluted tenfold with DEPC-treated water. For RT–PCR, EzWay Taq PCR MasterMix (Komabiotech) and 5 μl of synthesized cDNA as template were used. Thermal cycling was carried out under the following conditions: incubation at 95 °C for 1 min followed by 30 cycles of 30 s at 95 °C, 30 s at 55 °C, and 30 s at 72 °C. The primer pairs used in this experiment were designed so that the amplification product would include the breakpoints of the fusion genes. PCR products were analyzed by agarose gel electrophoresis. The primers used are summarized in Supplementary Table 11.
URLs.
Broad Institute Firehose platform for TCGA data, http://gdac.broadinstitute.org/; AROMA for SNP6 data preprocessing, http://www.aroma-project.org/; Cancer Genomics Hub for TCGA raw data, https://cghub.ucsc.edu/.
Accession codes.
Additional samples (R094–R114) from the SMC cohort have been deposited in the European Genome-phenome Archive (EGA) under accession EGAS00001001800. Additional samples from the INCB cohort have been deposited in the Sequence Read Archive (SRA) under accession SRP074425. Array CGH data for patients from the MD Anderson cohort have been deposited in the Gene Expression Omnibus (GEO) under accession GSE63035.
References
Ricard, D. et al. Primary brain tumours in adults. Lancet 379, 1984–1996 (2012).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Frattini, V. et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 45, 1141–1149 (2013).
Brennan, C.W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013).
Mazor, T. et al. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell 28, 307–317 (2015).
Kim, J. et al. Spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 28, 318–328 (2015).
Kim, H. et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 25, 316–327 (2015).
Nowell, P.C. The clonal evolution of tumor cell populations. Science 194, 23–28 (1976).
Nordling, C.O. A new theory on cancer-inducing mechanism. Br. J. Cancer 7, 68–72 (1953).
Armitage, P. & Doll, R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br. J. Cancer 8, 1–12 (1954).
Sottoriva, A. et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 110, 4009–4014 (2013).
Gill, B.J. et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. USA 111, 12550–12555 (2014).
Johnson, B.E. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014).
Suzuki, H. et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 47, 458–468 (2015).
Trifonov, V., Pasqualucci, L., Tiacci, E., Falini, B. & Rabadan, R. SAVI: a statistical algorithm for variant frequency identification. BMC Syst. Biol. 7 (suppl. 2), S2 (2013).
Melamed, R.D., Wang, J., Iavarone, A. & Rabadan, R. An information theoretic method to identify combinations of genomic alterations that promote glioblastoma. J. Mol. Cell Biol. 7, 203–213 (2015).
Ciriello, G., Cerami, E., Sander, C. & Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 22, 398–406 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Stieglitz, E. et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet. 47, 1326–1333 (2015).
Hunter, C. et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 66, 3987–3991 (2006).
Yip, S. et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 15, 4622–4629 (2009).
Cahill, D.P. et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 13, 2038–2045 (2007).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Miyazono, K., Olofsson, A., Colosetti, P. & Heldin, C.H. A role of the latent TGF-β1–binding protein in the assembly and secretion of TGF-β1. EMBO J. 10, 1091–1101 (1991).
Sterner-Kock, A. et al. Disruption of the gene encoding the latent transforming growth factor-β binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 16, 2264–2273 (2002).
Mermel, C.H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Trifonov, V., Pasqualucci, L., Dalla Favera, R. & Rabadan, R. MutComFocal: an integrative approach to identifying recurrent and focal genomic alterations in tumor samples. BMC Syst. Biol. 7, 25 (2013).
Singh, D. et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337, 1231–1235 (2012).
Di Stefano, A.L. et al. Detection, characterization, and inhibition of FGFR-TACC fusions in IDH wild-type glioma. Clin. Cancer Res. 21, 3307–3317 (2015).
Hegi, M.E. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003 (2005).
Schneider, T.D. & Stephens, R.M. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 (1990).
Alexandrov, L.B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Sakellarios Zairis, H.K., Blumberg, A. & Rabadan, R. Moduli spaces of phylogenetic trees describing tumor evolutionary patterns. Lect. Notes Computer Sci. 8609, 528–839 (2014).
Vogelstein, B. et al. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 319, 525–532 (1988).
Yates, L.R. & Campbell, P.J. Evolution of the cancer genome. Nat. Rev. Genet. 13, 795–806 (2012).
Wang, J. et al. Tumor evolutionary directed graphs and the history of chronic lymphocytic leukemia. eLife 3, e02869 (2014).
Carter, S.L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
Roth, A. et al. PyClone: statistical inference of clonal population structure in cancer. Nat. Methods 11, 396–398 (2014).
Kuan, C.T., Wikstrand, C.J. & Bigner, D.D. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr. Relat. Cancer 8, 83–96 (2001).
Torres-García, W. et al. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics 30, 2224–2226 (2014).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Verhaak, R.G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010).
Anido, J. et al. TGF-β receptor inhibitors target the CD44high/Id1high glioma-initiating cell population in human glioblastoma. Cancer Cell 18, 655–668 (2010).
Han, J., Alvarez-Breckenridge, C.A., Wang, Q.E. & Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 5, 945–955 (2015).
Joseph, J.V., Balasubramaniyan, V., Walenkamp, A. & Kruyt, F.A. TGF-β as a therapeutic target in high grade gliomas—promises and challenges. Biochem. Pharmacol. 85, 478–485 (2013).
Kaminska, B., Kocyk, M. & Kijewska, M. TGF β signaling and its role in glioma pathogenesis. Adv. Exp. Med. Biol. 986, 171–187 (2013).
Patel, A.P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).
Massagué, J. TGFβ in cancer. Cell 134, 215–230 (2008).
Fakhrai, H. et al. Eradication of established intracranial rat gliomas by transforming growth factor β antisense gene therapy. Proc. Natl. Acad. Sci. USA 93, 2909–2914 (1996).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Kent, W.J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Khalili, J.S., Hanson, R.W. & Szallasi, Z. In silico prediction of tumor antigens derived from functional missense mutations of the cancer gene census. OncoImmunology 1, 1281–1289 (2012).
Olshen, A.B., Venkatraman, E.S., Lucito, R. & Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004).
Magi, A. et al. EXCAVATOR: detecting copy number variants from whole-exome sequencing data. Genome Biol. 14, R120 (2013).
Iyer, M.K., Chinnaiyan, A.M. & Maher, C.A. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics 27, 2903–2904 (2011).
Abate, F. et al. Pegasus: a comprehensive annotation and prediction tool for detection of driver gene fusions in cancer. BMC Syst. Biol. 8, 97 (2014).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Niola, F. et al. Mesenchymal high-grade glioma is maintained by the ID–RAP1 axis. J. Clin. Invest. 123, 405–417 (2013).
Carro, M.S. et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325 (2010).
Zhao, X. et al. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 10, 643–653 (2008).
Acknowledgements
R.R. acknowledges funding from the NIH (U54 CA193313, R01 CA185486, and R01 CA179044). J.W. is supported by a Precision Medicine Fellowship (UL1 TR000040). E.L. is supported by the Cancer Biology Training Program (T32 CA09503). D.I.S.R. is supported by the US National Library of Medicine (T15 LM007079). S.Z. is supported by a Personalized Medicine Fellowship (TL1 TR000082). This work is also supported by NIH grants to A.L. (R01 CA101644 and R01 CA131126) and A.I. (R01 CA178546 and R01 NS061776 and a grant from the Chemotherapy Foundation). V.F. is supported by a fellowship from the American Brain Tumor Association (ABTA). G.F. is partly funded by the Italian Ministry of Health. D.-H.N. is supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C3418).
Author information
Authors and Affiliations
Contributions
E.C., M.E., and G.F. started the collection of samples and prepared libraries for genomic and transcriptomic analyses. E.C., V.F., and A.L. performed LTBP4 experiments. V.F. performed Sanger validation. S.Z. and A.J.B. performed the statistical analysis of phylogenetic trees. D.-H.N. provided additional data. F.A. and S.Z. ran Pegasus analysis. Z.L. ran ssGSEA analysis. D.I.S.R. constructed the mathematical model of tumor evolution before and after treatment. R.R., J.W., E.L., and O.E. developed and carried out analysis of genomic data. A.L., R.R., and A.I. conceived the methodology. D.-H.N., J.-K.L., I.-H.L., and W.-Y.P. provided clinical and genomic information for the SMC cohort. D.-H.N. and Y.-J.S. performed the validation of MGMT fusion. R.R., A.I., and J.W. conceived the project, discussed the results and implications, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–16 and Supplementary Note. (PDF 20648 kb)
Supplementary Data
Sanger validation of selected mutations. (PDF 32218 kb)
Supplementary Table 1
Coverage for whole-exome sequencing. (XLSX 67 kb)
Supplementary Table 2
All somatic mutations in patients with recurrent GBM. (XLSX 5494 kb)
Supplementary Table 3
Significant regions and potential driver genes reported by GISTIC2. (XLSX 150 kb)
Supplementary Table 4
Significant driver genes predicted by MutComFocal. (XLSX 366 kb)
Supplementary Table 5
Analysis of loss of heterozygosity. (XLSX 175 kb)
Supplementary Table 6
Selected list of gene fusion candidates. (XLSX 5925 kb)
Supplementary Table 7
Tumor purity estimation from ABSOLUTE. (XLSX 59 kb)
Supplementary Table 8
Cellular frequency prediction from PyClone. (XLSX 3816 kb)
Supplementary Table 9
EGFRvIII prediction from PRADA. (XLSX 53 kb)
Supplementary Table 10
Patient clinical data. (XLSX 29 kb)
Supplementary Table 11
Primer/oligo sequences for experimental validation. (XLSX 40 kb)
Supplementary Table 12
Validation of selected mutations by CancerSCAN. (XLSX 48 kb)
Supplementary Code
Glioblastoma evolution. (TXT 16 kb)
Rights and permissions
About this article

Cite this article
Wang, J., Cazzato, E., Ladewig, E. et al. Clonal evolution of glioblastoma under therapy. Nat Genet 48, 768–776 (2016). https://doi.org/10.1038/ng.3590
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3590
This article is cited by
-
Integrative multi-omics characterization reveals sex differences in glioblastoma
Biology of Sex Differences (2024)
-
Exploring fetal brain tumor glioblastoma symptom verification with self organizing maps and vulnerability data analysis
Scientific Reports (2024)
-
Glioblastoma immunotherapy: time for hope?
Neurological Sciences (2024)
-
Temporal and spatial stability of the EM/PM molecular subtypes in adult diffuse glioma
Frontiers of Medicine (2023)
-
Mutant-allele dispersion correlates with prognosis risk in patients with advanced non-small cell lung cancer
Journal of Cancer Research and Clinical Oncology (2023)
