
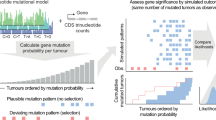
Abstract
Despite extraordinary efforts to profile cancer genomes, interpreting the vast amount of genomic data in the light of cancer evolution remains challenging. Here we demonstrate that neutral tumor evolution results in a power-law distribution of the mutant allele frequencies reported by next-generation sequencing of tumor bulk samples. We find that the neutral power law fits with high precision 323 of 904 cancers from 14 types and from different cohorts. In malignancies identified as evolving neutrally, all clonal selection seemingly occurred before the onset of cancer growth and not in later-arising subclones, resulting in numerous passenger mutations that are responsible for intratumoral heterogeneity. Reanalyzing cancer sequencing data within the neutral framework allowed the measurement, in each patient, of both the in vivo mutation rate and the order and timing of mutations. This result provides a new way to interpret existing cancer genomic data and to discriminate between functional and non-functional intratumoral heterogeneity.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Accession codes
References
Greaves, M. & Maley, C.C. Clonal evolution in cancer. Nature 481, 306–313 (2012).
Basanta, D. & Anderson, A.R.A. Exploiting ecological principles to better understand cancer progression and treatment. Interface Focus 3, 20130020 (2013).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Burrell, R.A., McGranahan, N., Bartek, J. & Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 501, 338–345 (2013).
Marusyk, A., Almendro, V. & Polyak, K. Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer 12, 323–334 (2012).
Polyak, K. Tumor heterogeneity confounds and illuminates: a case for Darwinian tumor evolution. Nat. Med. 20, 344–346 (2014).
Gerlinger, M. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 (2012).
Baca, S.C. et al. Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 (2013).
Tabassum, D.P. & Polyak, K. Tumorigenesis: it takes a village. Nat. Rev. Cancer 15, 473–483 (2015).
Shou, W., Bergstrom, C.T., Chakraborty, A.K. & Skinner, F.K. Theory, models and biology. eLife 4, e07158 (2015).
Sottoriva, A. et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 47, 209–216 (2015).
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Jesinghaus, M. et al. Distinctive spatiotemporal stability of somatic mutations in metastasized microsatellite-stable colorectal cancer. Am. J. Surg. Pathol. 39, 1140–1147 (2015).
Ohta, T. & Gillespie, J.H. Development of neutral and nearly neutral theories. Theor. Popul. Biol. 49, 128–142 (1996).
Donnelly, P. & Tavaré, S. Coalescents and genealogical structure under neutrality. Annu. Rev. Genet. 29, 401–421 (1995).
Durrett, R. & Schweinsberg, J. Approximating selective sweeps. Theor. Popul. Biol. 66, 129–138 (2004).
Driessens, G., Beck, B., Caauwe, A., Simons, B.D. & Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 488, 527–530 (2012).
Luria, S.E. & Delbrück, M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28, 491–511 (1943).
Griffiths, R.C. & Tavaré, S. The age of a mutation in a general coalescent. Communications in Statistics 14, 273–295 (1998).
Maruvka, Y.E., Kessler, D.A. & Shnerb, N.M. The birth-death-mutation process: a new paradigm for fat tailed distributions. PLoS One 6, e26480 (2011).
Durrett, R. Population genetics of neutral mutations in exponentially growing cancer cell populations. Ann. Appl. Probab. 23, 230–250 (2013).
Kessler, D.A. & Levine, H. Large population solution of the stochastic Luria-Delbruck evolution model. Proc. Natl. Acad. Sci. USA 110, 11682–11687 (2013).
Bak, P., Tang, C. & Wiesenfeld, K. Self-organized criticality: an explanation of the 1/f noise. Phys. Rev. Lett. 59, 381–384 (1987).
Jones, S. et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl. Acad. Sci. USA 105, 4283–4288 (2008).
Bozic, I. et al. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 107, 18545–18550 (2010).
Sun, S., Klebaner, F. & Tian, T. A new model of time scheme for progression of colorectal cancer. BMC Syst. Biol. 8 (suppl. 3), S2 (2014).
Helleday, T., Eshtad, S. & Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 15, 585–598 (2014).
Wang, K. et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 46, 573–582 (2014).
de Bruin, E.C. et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256 (2014).
Zhang, J. et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259 (2014).
Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012).
Attolini, C.S.-O. et al. A mathematical framework to determine the temporal sequence of somatic genetic events in cancer. Proc. Natl. Acad. Sci. USA 107, 17604–17609 (2010).
Gerstung, M., Eriksson, N., Lin, J., Vogelstein, B. & Beerenwinkel, N. The temporal order of genetic and pathway alterations in tumorigenesis. PLoS One 6, e27136 (2011).
Sprouffske, K., Pepper, J.W. & Maley, C.C. Accurate reconstruction of the temporal order of mutations in neoplastic progression. Cancer Prev. Res. (Phila.) 4, 1135–1144 (2011).
Guo, J., Guo, H. & Wang, Z. Inferring the temporal order of cancer gene mutations in individual tumor samples. PLoS One 9, e89244 (2014).
Sottoriva, A. et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 110, 4009–4014 (2013).
Wang, Y. et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512, 155–160 (2014).
Vermeulen, L. et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science 342, 995–998 (2013).
Heng, H.H.Q. et al. Stochastic cancer progression driven by non-clonal chromosome aberrations. J. Cell. Physiol. 208, 461–472 (2006).
Navin, N. et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011).
Marusyk, A. et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 514, 54–58 (2014).
Almendro, V. et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Reports 6, 514–527 (2014).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Koboldt, D.C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Andor, N. et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. doi:10.1038/nm.3984 (30 November 2015).
Niu, B. et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 30, 1015–1016 (2014).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Favero, F. et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 26, 64–70 (2015).
Acknowledgements
We thank D. Shibata, C. Curtis, S. Tavaré and R. Durrett for fruitful discussions. We would like to thank N. Andor (Stanford University) for supplying mutation calls for the TCGA data. We also thank V. Mustonen for useful suggestions.
A.S. is supported by The Chris Rokos Fellowship in Evolution and Cancer. B.W. is supported by the Geoffrey W. Lewis Post-Doctoral Training fellowship. This work was supported by the Wellcome Trust (105104/Z/14/Z). C.P.B. acknowledges funding from the Wellcome Trust through a Research Career Development Fellowship (097319/Z/11/Z). This work was supported by a Cancer Research UK Career Development Award to T.A.G. M.J.W. is supported by a UK Medical Research Council student fellowship.
This study makes use of data generated by the Department of Pathology of the University of Hong Kong and Pfizer, Inc.; a full list of the investigators who contributed to the generation of the data is available from ref. 28.
Author information
Authors and Affiliations
Contributions
M.J.W. and B.W. contributed to the development of the model. M.J.W. designed and performed computational simulations with support from C.P.B. M.J.W., A.S. and T.A.G. analyzed the data. C.P.B. contributed to the analysis. T.A.G. and A.S. jointly conceived, designed and developed the model, interpreted the results and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Rates of different mutation types in CRC.
(a) Stratification by mutation type indicates that C>T mutations occur at a significantly greater rate than other types. (b) As for the overall mutation rate (Fig. 1d), the rates of all mutation types were significantly higher in the MSI group.
Supplementary Figure 2 Analysis of neutral evolution in colon cancers is robust to copy number changes.
We removed mutations that fell within regions of the genome with altered copy number, that were identified by using SNP arrays paired to the exome-sequenced samples to detect regions of copy number change. (a) The consistently high values of goodness of fit demonstrate that the neutral model is robust to confounding copy number changes. Red line indicates the R2=0.98 threshold for calling a tumor neutral. (b) Estimating the mutation rates using only the mutations in copy number–devoid regions yields similar results as when all SNVs are included, confirming the robustness of our approach.
Supplementary Figure 3 Validation of the mutational signature across the frequency spectrum.
The mutational signature that characterizes the underlying biology is maintained across the frequency spectrum, providing further evidence that the identified somatic variants are reliable and are not due to sequencing errors.
Supplementary Figure 4 Synonymous/nonsynonymous ratio in colon cancers.
(a) A random base change within a codon is more likely to result in a nonsynonymous or stop-gain mutation than a synonymous mutation; hence, we expect the mutation rate per division of nonsynonymous mutations to be higher than for synonymous mutations. This is observed in the data. (b) The synonymous and nonsynonymous mutation rates become equivalent after normalizing by the total number of possible synonymous and nonsynonymous mutation sites in the exome, respectively. Therefore, synonymous and nonsynonymous mutations accrue at the same effective rate, consistent with our neutral model of cancer growth.
Supplementary Figure 5 Neutrality analysis in WGS gastric cancers data is robust to copy number changes.
(a) The goodness of fit of the neutral model was robust to copy number changes, as results were equivalent when only diploid regions or all genomic regions were considered. We note that the total number of cases considered after CNV filtering is considerably smaller because some samples did not have enough variants in the remaining diploid regions to fit the model. Red line indicates the R2=0.98 threshold for calling a tumor neutral. (b) Mutation rates were also consistent when copy number–altered regions were discarded in the analysis. We found only a single neutral MSI tumor, so a distribution could not be plotted.
Supplementary Figure 6 Validation of the mutational signature across the frequency spectrum in WGS gastric cancer data.
The mutational signature is conserved through the allelic frequency spectrum also in this cohort of whole genome–sequenced gastric cancers, thus supporting the authenticity of the called variants.
Supplementary Figure 7 Synonymous/nonsynonymous ratio in whole genome–sequenced gastric cancers.
Adjusted synonymous versus nonsynonymous mutation rates were consistent with neutral evolution in the gastric cancer cohort.
Supplementary Figure 8 Mutation rates by channel across cancer types.
When measured separately between different mutational channels, the mutation rate estimated by the model was consistent with the underlying biology: rates of C>A mutations were higher in lung cancer (tobacco smoking) and C>T mutation rates were higher than other channels in all cancer types.
Supplementary Figure 9 Stochastic simulations of neutral evolution recapitulate the observed NGS data.
(a) We produced realistic synthetic NGS data using a stochastic simulation of tumor growth that accounts for the neutral accumulation of mutations in the tumor as well as the different sources of sequencing noise (sampling, sequencing depth and normal contamination). (b) The prediction of the analytical model on the cumulative distribution of subclonal allelic frequencies agrees with the stochastic simulation. We generated synthetic data to test the accuracy with which tumor parameters could be reliably recovered when faced with confounding factors. Illustrative synthetic data are shown for (c) low mutation rate, (d) a high number of clonal mutations, (e) significant normal contamination and (f) a low detection limit. (g) Over 10,000 simulations, the interquartile range of the percentage error in the estimates of the mutation rate is <5%, demonstrating the ability of the analytical model to accurately estimate tumor growth parameters from NGS data. (h) The R2 values of the fits are consistently high over 10,000 simulations. Unless otherwise stated, the input parameters for the simulation and subsequent sampling were μ=100 mutations/cell division, λ=ln(2), detection limit=10%, normal contamination=0%, mean Ni=100 and number of clonal mutations=200.
Supplementary Figure 10 Robustness of the parameter estimation analysis.
By varying the parameters of the simulation, we show that the analytical model can accurately identify neutrality of tumor growth and recover the mutation rate in the face of (a) different numbers of clonal mutations and (b) different detection limits, and that we can correct for normal contamination accurately (to within 5%) for contamination below 30% (c,d). (e,f) The effect of varying read depth. Less accurate mutation rate estimates were achieved at low read depth (<25) and poorer fits of the analytical model. (g,h) The effect of mutation rate: lower mutation rates lead to poorer model fits and a higher variance in the mutation estimate because fewer variants are available to fit the model. (i,j) The effect of growth rate λ: the variance in the mutation rate estimate increases as tumor growth slows (l decreases) and the fit of the model becomes worse. The offspring probability distributions for the different values of λ were Pλ= ln(2) = (p0 = 0, p1 = 0, p2 = 1), Pλ = ln(1.8) = (p0 = 0.05, p1 = 0.1, p2 = 0.85), Pλ= ln(1.6) = (p0 = 0.1, p1 = 0.2, p2 = 0.7), Pλ= ln(1.4) = (p0 = 0.2, p1 = 0.2, p2 = 0.6) and Pλ = ln(2) = (p0 = 0.2, p1 = 0.4, p2 = 0.4).
Supplementary Figure 11 Clonal expansions and microenvironmental niches produce a deviation from the power-law.
By introducing a second population with a 65% fitness advantage (P = (p0 = 0, p1 = 0.8, p2 = 0.2), Q = (p0 = 0, p1 = 0.02, p2 = 0.98)) when the tumor comprises 80 cells, we see a second peak at VAF ~0.2 (a) and a bend in the cumulative distribution plot (b). A subclonal tumor architecture where mutations from the same subclone cluster around allelic frequencies would not show patterns consistent with neutrality, both when we consider two (c,d) and three (e,f) different subclones within a sample. A new phenotypically distinct clone introduced with a tenfold higher mutation rate (20 per division to 200 per division) also produces a deviation from the neutral power-law (g,h).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–11. (PDF 1799 kb)
Supplementary Data Set 1: Pan-cancer whole-exome data set.
Table of analyzed pan-cancer samples from TCGA. (XLS 399 kb)
Supplementary Data Set 2: Gastric cancer whole-genome data set.
Table of analyzed gastric cancer samples from Wang et al. (XLS 87 kb)
Rights and permissions
About this article

Cite this article
Williams, M., Werner, B., Barnes, C. et al. Identification of neutral tumor evolution across cancer types. Nat Genet 48, 238–244 (2016). https://doi.org/10.1038/ng.3489
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3489
This article is cited by
-
A statistical method for quantifying progenitor cells reveals incipient cell fate commitments
Nature Methods (2024)
-
Priors, population sizes, and power in genome-wide hypothesis tests
BMC Bioinformatics (2023)
-
State-dependent evolutionary models reveal modes of solid tumour growth
Nature Ecology & Evolution (2023)
-
Neuroblastoma arises in early fetal development and its evolutionary duration predicts outcome
Nature Genetics (2023)
-
Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity
Nature Genetics (2023)
