
Abstract
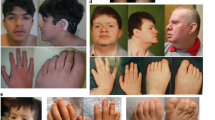
Temple-Baraitser syndrome (TBS) is a multisystem developmental disorder characterized by intellectual disability, epilepsy, and hypoplasia or aplasia of the nails of the thumb and great toe1,2. Here we report damaging de novo mutations in KCNH1 (encoding a protein called ether à go-go, EAG1 or KV10.1), a voltage-gated potassium channel that is predominantly expressed in the central nervous system (CNS), in six individuals with TBS. Characterization of the mutant channels in both Xenopus laevis oocytes and human HEK293T cells showed a decreased threshold of activation and delayed deactivation, demonstrating that TBS-associated KCNH1 mutations lead to deleterious gain of function. Consistent with this result, we find that two mothers of children with TBS, who have epilepsy but are otherwise healthy, are low-level (10% and 27%) mosaic carriers of pathogenic KCNH1 mutations. Consistent with recent reports3,4,5,6,7,8, this finding demonstrates that the etiology of many unresolved CNS disorders, including epilepsies, might be explained by pathogenic mosaic mutations.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Change history
06 February 2015
In the version of this article initially published, in Figure 2a, the order of the protein alterations for variants c.1465C>T and c.1480A>G was inverted. The correct protein alterations for these two variants are p.Leu489Phe and p.Ile494Val, respectively. This error has been corrected in the HTML and PDF versions of the article.
References
Temple, I.K. & Baraitser, M. Severe mental retardation and absent nails of hallux and pollex. Am. J. Med. Genet. 41, 173–175 (1991).
Gabbett, M.T., Clark, R.C. & McGaughran, J.M. A second case of severe mental retardation and absent nails of hallux and pollex (Temple-Baraitser syndrome). Am. J. Med. Genet. 146A, 450–452 (2008).
Depienne, C. et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J. Med. Genet. 47, 404–410 (2010).
Marini, C., Mei, D., Helen Cross, J. & Guerrini, R. Mosaic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia 47, 1737–1740 (2006).
Selmer, K.K. et al. Parental SCN1A mutation mosaicism in familial Dravet syndrome. Clin. Genet. 76, 398–403 (2009).
Kato, M. et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 54, 1282–1287 (2013).
Weckhuysen, S. et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25 (2012).
Jamuar, S.S. et al. Somatic mutations in cerebral cortical malformations. N. Engl. J. Med. 371, 733–743 (2014).
Yesil, G., Guler, S., Yuksel, A. & Alanay, Y. Report of a patient with Temple-Baraitser syndrome. Am. J. Med. Genet. 164A, 848–851 (2014).
Jacquinet, A. et al. Temple-Baraitser syndrome: a rare and possibly unrecognized condition. Am. J. Med. Genet. 152A, 2322–2326 (2010).
Adzhubei, I.A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Kumar, P., Henikoff, S. & Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Hemmerlein, B. et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol. Cancer 5, 41 (2006).
Urrego, D., Tomczak, A.P., Zahed, F., Stühmer, W. & Pardo, L.A. Potassium channels in cell cycle and cell proliferation. Phil. Trans. R. Soc. Lond. B 369, 20130094 (2014).
Gómez-Varela, D. et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 67, 7343–7349 (2007).
Zhang, J. et al. Regulation of cell proliferation of human induced pluripotent stem cell–derived mesenchymal stem cells via ether-à-go-go 1 (hEAG1) potassium channel. Am. J. Physiol. Cell Physiol. 303, C115–C125 (2012).
Ouadid-Ahidouch, H. & Le Bourhis, X. Changes in the K+ current-density of MCF-7 cells during progression through the cell cycle: possible involvement of a h-ether.a-gogo K+ channel. Recept. Channels 7, 345–356 (2000).
Petrovski, S., Wang, Q., Heinzen, E.L., Allen, A.S. & Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 9, e1003709 (2013).
Tian, C. et al. Potassium channels: structures, diseases, and modulators. Chem. Biol. Drug Des. 83, 1–26 (2014).
Ufartes, R. et al. Behavioural and functional characterization of KV10.1 (Eag1) knockout mice. Hum. Mol. Genet. 22, 2247–2262 (2013).
D'Adamo, M.C., Catacuzzeno, L., Di Giovanni, G., Franciolini, F. & Pessia, M. K+ channelepsy: progress in the neurobiology of potassium channels and epilepsy. Front. Cell Neurosci. 7, 134 (2013).
Barcia, G. et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 44, 1255–1259 (2012).
Heron, S.E. et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 44, 1188–1190 (2012).
Browne, D.L. et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat. Genet. 8, 136–140 (1994).
Lee, Y.-C. et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 72, 859–869 (2012).
Coucke, P.J. et al. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum. Mol. Genet. 8, 1321–1328 (1999).
Tawil, R. et al. Andersen's syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 35, 326–330 (1994).
Epi4K Consortium & Epilepsy Phenome/Genome Project. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013).
Poduri, A., Evrony, G.D., Cai, X. & Walsh, C.A. Somatic mutation, genomic variation, and neurological disease. Science 341, 1237758 (2013).
Cleary, J.G. et al. Joint variant and de novo mutation identification on pedigrees from high-throughput sequencing data. J. Comput. Biol. 21, 405–419 (2014).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Simons, C. et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Hum. Genet. 92, 767–773 (2013).
Bolger, A.M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Koboldt, D.C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Schroeder, C.I. et al. Chemical synthesis, 3D structure, and ASIC binding site of the toxin mambalgin-2. Angew. Chem. Int. Edn Engl. 53, 1017–1020 (2014).
Garg, V., Sachse, F.B. & Sanguinetti, M.C. Tuning of EAG K+ channel inactivation: molecular determinants of amplification by mutations and a small molecule. J. Gen. Physiol. 140, 307–324 (2012).
Acknowledgements
We would like to thank the patients and their families for their participation in this study. Computational support was provided by the NeCTAR Genomics Virtual Laboratory and QRIScloud programs. We thank the Queensland Centre for Medical Genomics and Institute for Molecular Bioscience sequencing facility teams for their assistance with this project and J. Lynch for providing access to his patch-clamp facilities. We also wish to thank J. Vandenberg for his helpful discussion of results. R.J.T. was supported by an Australian Research Council Discovery Early-Career Research Award. L.M. is supported by an Australian National Health and Medical Research Council (NHMRC) C.J. Martin fellowship. A.J. and F.-G.D. were supported by a Fund Invest for Scientific Research (FIRS) grant from Centre Hospitalier Universitaire of Liége, Belgium. This work was supported in part by a University of Queensland Foundation Research Excellence Award and a donation by Joan and Graham J. Crawford.
Author information
Authors and Affiliations
Contributions
C.S., R.J.T., M.T.G. and J.M. conceived and designed the project. M.T.G., J.M., Y.A., A.J., F.-G.D., A.V., J.S., G.Y., S.G. and A.Y. phenotyped the cases, provided clinical samples and provided clinical information. D.M. and S.M.G. were responsible for exome sequencing. C.S. performed all exome and variant analysis, with input from R.J.T. J.G.C. provided software and advice for sequence analysis. J.C., B.C.-A. and K.R. performed Sanger sequencing and constructed the expression vectors. L.D.R., L.M. and G.F.K. conceived the electrophysiology experiments, which were performed by L.D.R., B.C.-A. and L.M. J.C. prepared samples for targeted amplicon sequencing. Amplicon data analysis was performed by C.S. and G.J.B. All data were reviewed and synthesized by C.S. and R.J.T. The manuscript was drafted by C.S. and R.J.T. with input from M.T.G. and L.D.R. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
R.J.T. became an employee of Illumina, Inc., during the course of this project.
Integrated supplementary information
Supplementary Figure 1 Activation, deactivation and Inactivation of wild-type and mutant KCNH1 channels expressed in Xenopus oocytes.
(a) Representative families of whole-cell currents showing voltage-dependent activation of wild-type (WT) and mutant human KCNH1 channels expressed in Xenopus oocytes. Currents were elicited by steps from –120 mV to +80 mV in 20-mV increments, using a holding potential of –100 mV. Inset in the final panel: schematic of the pulse protocol used for the activation experiments. (b) Representative families of whole-cell currents showing only minor steady-state inactivation of WT and mutant KCNH1 channels expressed in Xenopus oocytes. Inset in the final panel: schematic of the pulse protocol used for the inactivation experiments. (c) Activation current-voltage relationship (± s.e.m.) for WT, p.Lys217Asn, p.Leu489Phe, p.Ile494Val and p.Gln503Arg channels expressed in Xenopus oocytes. Currents were measured at the end of the activation test pulse. (d) Fast and slow time constants (τ) of deactivation for WT and mutant KCNH1 channels expressed in Xenopus oocytes, analyzed from the tail current at –90 mV following a maximally activating prepulse to +80 mV. (e) Steady-state inactivation current-voltage relationship (± s.e.m.) for WT and mutant KCNH1 channels expressed in Xenopus oocytes. Currents were measured at the end of the +30 mV activation test pulse. Data are presented as mean ± s.e.m. with the numbers of experiments indicated in parentheses. P values were calculated in comparison to WT using an unpaired t test with Welch’s correction. *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary information
Supplementary Text and Figures
Supplementary Figure 1, Supplementary Tables 1–3 and Supplementary Note. (PDF 681 kb)
Source data
Rights and permissions
About this article

Cite this article
Simons, C., Rash, L., Crawford, J. et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet 47, 73–77 (2015). https://doi.org/10.1038/ng.3153
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3153
This article is cited by
-
Overexpression of KCNN4 channels in principal neurons produces an anti-seizure effect without reducing their coding ability
Gene Therapy (2024)
-
Establishment and characterization of ZJUCHi003: an induced pluripotent stem cell line from a patient with Temple–Baraitser/Zimmermann–Laband syndrome carrying KCNH1 c.1070G > A (p.R357Q) variant
Human Cell (2024)
-
14-3-3 proteins regulate cullin 7-mediated Eag1 degradation
Cell & Bioscience (2023)
-
Mechanism underlying delayed rectifying in human voltage-mediated activation Eag2 channel
Nature Communications (2023)
-
Intracellular hemin is a potent inhibitor of the voltage-gated potassium channel Kv10.1
Scientific Reports (2022)
