
Abstract
Colobines are a unique group of Old World monkeys that principally eat leaves and seeds rather than fruits and insects. We report the sequencing at 146× coverage, de novo assembly and analyses of the genome of a male golden snub-nosed monkey (Rhinopithecus roxellana) and resequencing at 30× coverage of three related species (Rhinopithecus bieti, Rhinopithecus brelichi and Rhinopithecus strykeri). Comparative analyses showed that Asian colobines have an enhanced ability to derive energy from fatty acids and to degrade xenobiotics. We found evidence for functional evolution in the colobine RNASE1 gene, encoding a key secretory RNase that digests the high concentrations of bacterial RNA derived from symbiotic microflora. Demographic reconstructions indicated that the profile of ancient effective population sizes for R. roxellana more closely resembles that of giant panda rather than its congeners. These findings offer new insights into the dietary adaptations and evolutionary history of colobine primates.
Similar content being viewed by others


Main
Knowledge of the patterns and processes underlying the evolution of alternative dietary strategies in nonhuman primates is critical to understanding hominin evolution, nutritional ecology and applications in biomedicine1. Colobines, a group of Old World monkeys, serve as an important model organism for studying the evolution of the primate diet because of their adaptation to folivory: they primarily eat leaves and seeds rather than fruits and insects as their major food source. In their specialized and compartmentalized stomachs, colobines allow symbiotic bacteria in the foregut to ferment structural carbohydrates and then recover nutrients by digesting the bacteria2. This strategy is similar to that used by other foregut fermenters found in an evolutionarily distantly related group of mammals (for example, artiodactyls). Although a number of primate genomes have been sequenced thus far, high-quality genome sequence information is absent for Asian and African colobines, a key group for elucidating the evolution and adaptation of primates as a whole. Snub-nosed monkeys (Rhinopithecus species) are a group of endangered colobines, which were once widely distributed in Asia but are now limited to mountain forests in China and Vietnam3 (Supplementary Fig. 1). The golden snub-nosed monkey (GSM, R. roxellana) is recognized as an iconic endangered species in China for its golden coat, blue facial coloration, snub nose and specialized life history. Among its congeners, the black-white snub-nosed monkey (R. bieti), endemic to the Tibetan plateau, has the highest altitudinal distribution (>4,000 m above sea level) of any nonhuman primate. Given the above features and the fact that Rhinopithecus species consume difficult-to-digest foods that contain tannins (for example, leaves and pine seeds), we expected to identify genetic adaptations that enhance the breakdown of toxins, improve the regulation of energy metabolism and facilitate the digestion of symbiotic microbacteria.
Results
Genomic sequences and the accumulation of Alu sites
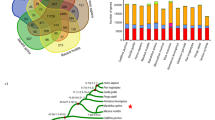
We sequenced the genome of a male GSM using a whole-genome shotgun strategy. Cleaned-up data provided 146-fold average coverage across 3.05 Gb of assembled sequence (Supplementary Fig. 2 and Supplementary Tables 1 and 2), with N50 values of 25.5 kb and 1.55 Mb, respectively, for contigs and scaffolds in the final assembly (generated using SOAPdenovo4; Supplementary Figs. 3,4,5 and Supplementary Tables 3–5). We identified a total of 21,813 protein-coding genes (94.52% of which were functionally classified; Fig. 1a, Supplementary Figs. 6,7,8 and Supplementary Tables 6 and 7). Of these gene annotations, 89.7% were supported by evidence of transcription (Supplementary Tables 8 and 9). Using these protein-coding genes, we generated a timescale for primate evolution (Fig. 1b and Supplementary Note).

(a) Venn diagram of shared orthologous gene families in GSM and six additional primate species. (b) Phylogenetic tree and estimated divergence times for GSM and other mammals. The outside edges for the shaded gray, purple and blue areas represent the boundaries for the Paleogene-Cretaceous, Cretaceous-Neogene and Miocene to the present, respectively.
Mobile elements comprised nearly half of the GSM genome (Supplementary Fig. 9 and Supplementary Tables 10–13). There were roughly 3,054 and 3,311 unique insertions of long interspersed element 1 (LINE1; L1) in GSM and rhesus macaque, respectively, more than in human (2,365 new insertions) and chimpanzee (1,841 new insertions) (Fig. 2a and Supplementary Table 14). However, the numbers of lineage-specific SVA (SINE-R, VNTR and Alu-like) elements were generally lower in Old World monkeys (nearly 0 specific inserts) than in the Hominoidea (855 and 294 new inserts for human and chimpanzee, respectively). Alu elements (a family of primate-specific repeats) were more dynamic in Old World monkeys than in human and chimpanzee, with approximately 55,972 and 43,381 unique recent insertions in GSM and rhesus macaque, respectively (Fig. 2a).

(a) Lineage-specific L1, SVA and Alu insertions in Old World monkeys and Hominoidea (apes and humans); mya, million years ago. (b) Enrichment for PSGs, genes evolved in parallel and expanded gene families that function in fatty acid and energy metabolism. The PSGs in GSM are shown in purple, the genes from expanded families in GSM are shown in light blue and the genes evolved in parallel in GSM and cattle are shown in orange. The sequence alignment shows sites that evolved in parallel in ACADSB, and the amino acids associated with each node represent the ancestral amino acids. (c) An unrooted neighbor-joining tree constructed using 33 olfactory receptor genes identified as REPs in GSM and cattle. Annotations in different colors denote the potential odorant recognition of the olfactory receptor genes.
Along with shaping genome diversity, abundant independently inserted elements can also contribute to nonallelic homologous recombination events that result in copy number variation and disease5. Reconstruction of the ancestral genome of catarrhini using the inferCARs algorithm6 identified 10 interchromosomal and 53 intrachromosomal breaks in the GSM lineage in comparison to 20 interchromosomal and 50 intrachromosomal breaks in rhesus macaque and 5 interchromosomal and 22 intrachromosomal breaks in human (Supplementary Fig. 10). Given that the scaffolds for the GSM assembly had not been assigned to chromosomes, the number of breaks estimated in GSM is expected to be an underestimate. To investigate the properties of these breakpoints, we explored 50-kb intervals centered on the ends of each conserved segment where the adjacent segments in the ancestor and target species differed. As in the mouse and human genomes6, the breakpoint regions in GSM were enriched for protein-coding genes (13.80 genes/Mb in breakpoint regions versus 7.64 genes/Mb in the whole genome), segmental duplication regions (12.78 in breakpoint regions versus 1.43 in the whole genome) and especially for repeat elements (Supplementary Table 15), suggesting that the occurrence of chromosome breakage in mammalian genomes is not random.
We did not find significant differences in the number of events of gene family expansion (P = 0.18, Mann-Whitney U test) or contraction (P = 0.88, Mann-Whitney U test) in comparisons of the Old World monkeys and Hominoidea. The rate of processed pseudogene formation, which requires the same functional L1 machinery as Alu insertion7, was similar for the GSM (15.4 per million years) and human (15.1 per million years) genomes (Supplementary Fig. 11 and Supplementary Table 16). Overall, the enrichment of Alu repeats in Old World monkeys did not appear to lead to significantly more rearrangements and changes in gene family composition on a genome-wide scale.
Evolution of gene families and adaptation to leaf consumption
We analyzed the functional properties of the gene families that had undergone expansion in the GSM genome to identify potential evolutionary events that might be related to adaptations consistent with leaf consumption. The GSM genome showed substantial expansion of gene families that are mainly involved in xenobiotic biodegradation, including thiamine metabolism (P < 0.01), lysosome (P < 0.01) and drug metabolism–other enzymes (P < 0.05) (all Fisher's exact test) (Supplementary Table 17). After removing orthologs in segmental duplication regions shared by GSM and rhesus macaque, we found that the GSM-specific genes embedded in segmental duplication regions were significantly enriched for pathways related to drug metabolism–other enzymes (P < 1 × 10−4), porphyrin and chlorophyll metabolism (P < 1 × 10−4), lysosome (P < 0.01) and drug metabolism–cytochrome P450 (P < 0.01) (all Fisher's exact test) (Supplementary Fig. 12 and Supplementary Tables 18–21). Genes enriched in the lysosome pathway encoded proteins belonging to the cysteine proteinase inhibitor superfamily (CST-3 to CST-7), which are abundant in salivary secretions8 and have been demonstrated to interact with tannic acid in the saliva of primates9. Thus, the expansion of salivary protein-coding genes and xenobiotic biodegradation genes would enhance the ability of colobines to detoxify secondary compounds present in leaves.
We identified 105 positively selected genes (PSGs) in the GSM lineage using the branch-site likelihood ratio test (Supplementary Table 22)10, including ones enriched for fatty acid pathways (P = 0.004, Fisher's exact test), which were mainly associated with the elongation and biosynthesis of fatty acids (Fig. 2b and Supplementary Table 23), as well as corresponding upstream and downstream pathways, i.e., signaling pathways for insulin (P = 0.01), adipocytokine (P = 0.01), and valine, leucine and isoleucine degradation (P = 0.005) (all Fisher's exact test) (Fig. 2b and Supplementary Table 23). Additionally, Gene Ontology (GO) terms related to fatty acids were also found to be over-represented by PSGs, including propanoate metabolism (P = 0.01), pyruvate metabolism (P = 0.04) and lipid binding (P = 0.03) (all Fisher's exact test) (Supplementary Table 24). These observations are consistent with a need to cope with the energy-rich, short-chain volatile fatty acids that are the main energy source of foregut fermenters (including GSM), produced during the process of degradation of plant cell wall components (celluloses and hemicelluloses)11. The foods most commonly eaten by GSM (such as Usnea diffracta Vain., leaves from Rosaceae and tree bark) are high-fiber, low-energy nutritional sources, and it therefore seems reasonable to hypothesize that leaf-eating monkeys living in high-altitude forests should employ enhanced energy metabolism to efficiently absorb and exploit scarce nutrients. For example, ACADM and HADNB encode the enzymes involved in catalyzing the initial and final reactions in the β-oxidation of fatty acids12,13. ALDH3A2, a typical fatty aldehyde dehydrogenase, catalyzes oxidation of the long-chain aldehydes produced by lipid metabolism. An absence of ALDH3A2 function contributes to Sjogren-Larsson syndrome, which typically involves ichthyosis, spastic diplegia and severe learning difficulties in humans14. Adaptive changes in the β-oxidation of fatty acids and lipid metabolism are expected to yield sufficient increases in the amount of ATP to guarantee that foregut fermenters have the required energy.
We identified 2,290 genes with significantly elevated rates of nonsynonymous substitution in both the GSM and cattle genomes (Supplementary Table 25), with these genes representing rapidly evolving proteins (REPs). Nearly half of the 105 PSGs in the GSM lineage were identified as REPs (49 PSGs; 46.7%) in both GSM and cattle. Interestingly, 198 REPs contained amino acid positions that showed parallel evolutionary patterns in both lineages (Supplementary Table 25), pointing to the existence of similar protein sequence substitutions in lineages that have independently acquired foregut fermentation. Intriguingly, 33 olfactory receptor proteins (Supplementary Tables 26 and 27) were identified as REPs. We identified potential target specificities for these 33 rapidly evolving olfactory receptors in GSM and cattle by comparing the amino acid sequences to those of receptors in human and mice with previously described information on odor perception. We found that 23 of 33 (69.7%) rapidly evolving olfactory receptors had been suggested to function in the perception of fruity, lemony and floral/woody odors (Fig. 2c, Supplementary Figs. 13 and 14, and Supplementary Table 27), consistent with plant-based odor perception.
It is well documented that there is a higher proportion of olfactory receptor pseudogenes in hominoids and Old World monkeys (the vision priority hypothesis) because their complete trichromatic color vision system is hypothesized to negate the need for high olfactory sensitivity15. We found that more than half of the olfactory receptor genes in GSM were pseudogenes or gene fragments (360 of 583; 61.7%) (Supplementary Figs. 15,16,17,18,19 and Supplementary Table 28), a proportion that is higher than for other primates and mammals16. The reduction in the olfactory system for GSM might have coincided with the recession of the external nostril in this species, as olfactory receptors are known to be expressed on the cilia of olfactory sensory neurons of the neuroepithelium in the nasal cavity.
New insight into the functional evolution of RNASE1
Convergent evolution of pancreatic RNase (encoded by RNASE1) in the colobines and ruminants is thought to be a response to the necessity for digesting high concentrations of bacterial RNA derived from the symbiotic microflora in the stomach required to recover nutrients17. Duplicated RNASE1 genes have been described as potentially enhancing digestive efficiency at low pH in the leaf monkeys Pygathrix nemaeus and Colobus guereza17,18. We assembled the two copies of the GSM pancreatic RNase gene in one scaffold and found the duplication segment containing RNASE1 to be approximately 34 kb in length (Fig. 3a), forming a tandem array with the original copy. We successfully amplified the border regions of the duplicates in eight colobine genera (Rhinopithecus, Pygathrix, Nasalis, Semnopithecus, Trachypithecus, Presbytis, Colobus and Piliocolobus), but these regions were not present in human and rhesus macaque (Supplementary Fig. 20 and Supplementary Table 29), suggesting that all colobine duplicates are derived from a single event in the ancestor of extant colobines. Phylogenetic trees reconstructed using both the coding sequences (468 bp) and flanking noncoding sequences (1,575 bp) from these genera showed no signs of gene conversion in noncoding regions (Supplementary Table 30). Thus, most signals of gene conversion within coding regions are likely to have undergone an adaptive sweep, as the homogenization of the coding region would potentially be harmful in functional divergence19 (Supplementary Figs. 21 and 22).

(a) Gene structures of RNASE1 and RNASE1B in GSM. The red and green lines denote the two duplication regions. Genes are indicated as unfilled boxes, and read depths are presented. TF and TR represent primers used to amplify the border regions of duplicates from eight colobine genera. (b) Phylogram of colobine pancreatic RNase proteins. Parallel adaptive substitutions from a basic amino acid to a neutral amino acid at residue 39 are labeled for the clades where they took place. Basic amino acids and neutral amino acids are marked in red and blue, respectively. Seven previously inferred parallel amino acid substitution sites are also given. Amino acids identical to those in human RNASE1 are indicated by dots. pI, isoelectric point. (c) Enzymatic activity of RNase against yeast tRNA at various pH levels. ANB refers to the ancestral protein of all Asian colobine RNASE1B proteins. ANB Arg39Trp refers to recombinant protein with the p.Arg39Trp alteration. Three technical replicates were performed for each data point, and the associated 95% confidence intervals are given. (d) Expression of RNASE1, RNASE1B and ACTB by quantitative RT-PCR in five different tissues of GSM. Three technical replicates were performed for each data point, and the associated 95% confidence intervals are presented.
In addition to RNASE1 and its duplications, we found an 810-bp processed pseudogene on a different scaffold. This pseudogene existed in all Asian colobine genomes but was absent from the African colobine genomes, suggesting that the retrotransposition event occurred after the African-Asian colobine split but before the divergence of the extant Asian genera (Fig. 3b). We expressed recombinant RNASE1 and RNASE1B proteins from R. roxellana, Presbytis melalophos and Trachypithecus francoisi to examine their ribonucleolytic activities at different pH levels. We found the same optimal pH at 7.4 for the RNASE1 proteins but an optimal pH ranging from 6.4 to 6.6 for the RNASE1B proteins from all three genera, suggesting that a lower optimal pH for RNASE1B is universal in colobines. Seven parallel amino acid changes (p.Arg4Gln, p.Lys6Glu, p.Arg39Trp, p.Pro42Ser, p.Arg78Lys, p.Arg98Gln and p.Ala122Asp) are suggested to have caused a decrease in the optimal pH for RNASE1B18,20; however, in our study with a larger sampling of genera, these loci were no longer conserved in the RNASE1 and RNASE1B proteins, indicating that none of these amino acids are required for reduction in the optimal pH level. We found a radical replacement at residue 39 of RNASE1B in all colobines, from a basic to a non-basic amino acid in comparison to the conserved basic amino acid (arginine) at this position in the RNASE1 proteins of all colobines and other primates. We reconstructed the Asian colobine ancestral RNASE1B gene (ANB) and expressed it in Escherichia coli. The optimal pH of the encoded ancestral protein was 7.0, falling between the values for the RNASE1 and RNASE1B proteins. We also mutated the ancestral gene to encode a neutral rather than basic amino acid (Arg39Trp) and determined that the optimal pH for this enzyme was 6.4, within the pH range (6.3–6.6) of the RNSAE1B proteins from Asian colobines (Fig. 3c). These results suggested that residue 39 is key in decreasing the optimal pH from 7.0 to 6.4. Furthermore, substitution at this site appears to have taken place in parallel on at least three occasions, once in the African (p.Arg39Trp) and twice in Asian (p.Arg39Trp and p.Arg39Gln) colobine clades. We also measured the expression level of each copy across five GSM tissues (pancreas, liver, spleen, kidney and muscle) and only found a noticeable difference in the expression of the two copies in the pancreas, where RNASE1B exhibited ∼80-fold upregulation in comparison to RNASE1 (Fig. 3d). These results imply that the two genes might be under the control of different regulatory regions and have different requirements for expression.
Stomach metagenomics and cellulose metabolism
To investigate the genetic bases of digestion of cellulose and hemicellulose, we examined the microbial communities of the GSM stomach using bacterial 16S rRNA next-generation sequencing. A total of 64,406 reads were clustered at 97% sequence similarity, representing 4,636 operational taxonomic units (Supplementary Table 31), indicating that the GSM microbiota are similar to those in human gastric fluid21, being dominated by Firmicutes (38.7%), Proteobacteria (28.9%) and Bacteroidetes (28.8%) but with more Proteobacteria (28.9% in GSMs versus 6.9% in human) and fewer Actinobacteria (1.1% in GSMs versus 10.7% in human) (Fig. 4). At the genus level, the GSM microbiota were similar to those from cattle rather than humans and were associated with fermentation (Supplementary Fig. 23, Supplementary Table 32 and Supplementary Note).

Relative abundance of microbial flora and taxonomic assignments from human, giant panda, cattle and GSM samples.
A de novo assembly of 10 Gb of RNA sequencing (RNA-seq) data from the GSM stomach generated a set of ∼88 Mb of contigs and 121,458 ORFs. The predicted ORFs were enriched in the metabolism of carbohydrates, amino acids, nucleotides, glycans and vitamins and especially in the metabolism of fatty acids (Supplementary Fig. 24 and Supplementary Table 33). This enrichment was further evidenced by the functional analysis of our data with metabolic terms focused mainly on energy production and conversion (Supplementary Fig. 25). We also identified many genes involved in the cellulose digestion pathway, including 27 cellulose genes, 17 1,4-β-cellobiosidase genes and 179 β-glucosidase genes. Glycoside hydrolase genes were also identified, and over 929 genes and modules were recovered from 47 different CAZy families (carbohydrate active enzymes). Although there were not significant differences in the abundance of debranching enzymes in comparisons of the GSM, giant panda22, tammar23 and termite24 hindguts, there were considerably more GH78 sequences in GSM. As in giant panda, the abundance of cellulases and endohemicellulases in GSM was generally lower than in human, cattle and termite (Supplementary Table 34).
Genomic variation and demographic history
Five species of snub-nosed monkeys are currently recognized in the genus Rhinopithecus, with three of them (R. roxellana, R. brelichi and R. bieti) endemic to China. The phylogenetic relationships within this group are controversial25, so we used short-read sequencing to address this question from a genome-wide perspective, carrying out whole-genome sequencing of the black-white (R. bieti), gray (R. brelichi) and Myanmar (R. strykeri) snub-nosed monkeys with approximately 30-fold coverage for each individual (∼282 Gb in total) (Supplementary Fig. 26 and Supplementary Tables 35–38). Using these reads, we identified 16.5 million putative variants among the 4 species. The rates at which heterozygous variants occurred indicated that the level of genetic diversity among the species of snub-nosed monkeys was much lower than for all other reported non-human primates (0.02–0.07% in comparison to 0.08–0.24%)26,27, with R. brelichi showing the highest value (0.07%), which approaches the human value (0.066%). The proportions of autosomal regions of homozygosity (ROHs) also supported the differences in heterozygous SNPs among snub-nosed monkeys (Fig. 5a and Supplementary Table 39). The heterozygosity in R. roxellana was higher than in R. bieti and R. strykeri, coinciding with its wider distribution and largest contemporary population size among the five snub-nosed monkey species28. However, the higher heterozygosity of R. brelichi is in contrast to its low population size (fewer than 800 individuals29) in comparison to other snub-nosed monkeys in the same genus.

(a) Model showing the phylogenetic relationships and speciation (left), heterozygosity (middle) and ROHs (right) among snub-nosed monkeys. Each 'violin' contains a vertical black line (25–75% range) and a white point inside (median), with the width depicting a kernel density trace rotated by 90° and its reflection. m, migration. Gray background shading denotes the heterozygosity range of nonhuman primates. (b) Demographic histories of snub-nosed monkeys reconstructed using the PSMC model. The Ne of giant panda is rescaled. The gray dashed line shows the MAR of Chinese loess. g (generation time) = 10 years; μ (neutral mutation rate per generation) = 1.36 × 10−8. (c) Heat map displaying the relative correlation of PSMC and MAR for snub-nosed monkeys. (d) Comparison of the historic (lighter shade) and current (darker shad) geographical locations of giant panda (orange) and GSM (purple).
To identify functional changes in the Rhinopithecus genome, we screened the mutations in orthologs across the four Rhinopithecus assemblies and compared the resulting changes to the human genome. A total of 1,010 Rhinopithecus amino acid alterations encoded in 824 genes were predicted to cause functional changes when analyzed in the context of their human counterparts (Supplementary Table 40). In 51 Rhinopithecus proteins, the observed amino acid variants were found to be disease causing or associated with disease according to the Human Gene Mutation Database (Supplementary Table 41). None of these variants were found at positions that are completely conserved in vertebrates, consistent with neutral theory expectations30.
Comparing orthologs between rhesus macaque and the snub-nosed monkeys, we reconstructed a maximum-likelihood phylogeny supporting the hypothesis of a separation between the northern species (R. roxellana and R. brelichi) and the 'Himalayan' species (R. bieti and R. strykeri)25 with 100% bootstrap support values (Fig. 5a). Molecular dating estimated that the split of the northern species occurred about 1.60 million years ago, immediately after the initiation of divergence among the four snub-nosed monkeys (1.69 million years ago). These times coincide with the uplift of the Tibetan plateau—Yuanmu movement (∼1.6 million years ago)—which helped to create the higher mountains of the Himalayas31 and might have resulted in the split of the northern and Himalayan species as well as the subsequent separation of R. roxellana and R. brelichi. Divergence within the Himalayan species appears to have begun about 0.3 million years ago, more recently than previously estimated25. The short branches of these lineages in the tree also imply that incomplete lineage sorting (ILS; cases where the gene tree was different from the species tree) and/or introgression have occurred within this clade. Using the coalescent hidden Markov model (CoalHMM)32,33, we quantified the amount of ILS as ∼5.8% of the genomes in the snub-nosed monkeys on the basis of all SNPs (regions where R. roxellana and R. brelichi were more closely related to R. bieti than to R. strykeri), a ratio less than that (∼30%) found in the genomes of great apes (human, chimpanzee and gorilla)27. CoalHMM was also employed to date the speciation events within this lineage, and the results supported a relatively recent isolation of snub-nosed monkeys: 0.15 ± 0.10 million years ago for the Himalayan species and 0.62 ± 0.17 million years ago for the northern species.
It is noteworthy that there were two distinct peaks in the distribution of speciation times for the Himalayan species, one close to the inferred genomic divergence time (0.3 million years ago) and the other within the speciation time (Supplementary Fig. 27), which coincided with the penultimate glaciation (0.13–0.3 million years ago)31. The relatively cold climate in this glaciation could potentially have caused isolation of R. bieti and R. strykeri. Historical fluctuations in effective population size (Ne) for the four snub-nosed monkeys were reconstructed with the help of the pairwise sequential Markovian coalescent (PSMC) model34, and two bottlenecks and two expansions were identified for R. roxellana (Fig. 5b and Supplementary Fig. 28). A similar pattern of historical trends has been found for giant panda35 (Spearman's r = 0.83, P < 0.01) (Fig. 5c), which is sympatric with R. roxellana. These concordant patterns suggest that mammals sharing the same habitant might feature a similar demographic history. The first major change in Ne was inferred to have occurred between 0.1 and ∼2 million years ago with a peak at ∼1 million years ago, close to the switch of the dominant wavelength for climate cycles from 41,000 to 100,000 years36. Population bottlenecks of Ne at this time have been identified in other mammals, such as giant panda35, brown bear37 and some great apes26, suggesting that global glaciations and severely cold climates at the Early-Middle Pleistocene boundary38 had substantial evolutionary impact on the population sizes of several large mammals. After the first bottleneck, the Ne for R. roxellana recovered and peaked at ∼0.07 million years ago (Fig. 5b). This recovery for R. roxellana was slightly different from that for giant panda, which was inferred to have occurred around 30,000–40,000 years ago. Although the precise extent of this disparity needs further validation, it offers a preliminary surmise that the relatively cold and dry climate of MIS (marine isotope stage) 4 (58,000–74,000 years ago), as indexed by the mass accumulation rate (MAR) of Chinese loess39, did not affect the effective population size of R. roxellana but did have a strong negative effect on giant panda. One potential explanation for the disparity is that giant panda evolved a highly specialized diet (bamboo) and recovered at relatively warm temperatures with expansion of alpine conifer forests35, whereas R. roxellana evolved a set of physiological traits that enabled it to tolerate a broader-based diet composed of difficult-to-digest foods, such as leaves, bark, lichen and seeds, as well as the ability to survive in high-altitude forests under conditions of hypoxia. It is also worth noting that the speleothem record of stalagmites in the mountain-surrounded Sichuan Basin of southwest China suggests the absence of a glacial maximum during MIS4 in this area40. Thus, more attention should be paid to the disparity between local and global climatic conditions in shaping the evolutionary history and biogeography of species in this region of Asia. A sharp decline in Ne for R. roxellana coincided with the extreme cooling climate during the last glaciation, and a similar decrease in Ne has been identified at this time in several other mammals26,35,37.
Historical trends of Ne for R. bieti, R. strykeri and R. brelichi showed a very different pattern from that of R. roxellana. A continuing decrease in Ne after divergence was found in these three species. This result may best be explained by habitat fragmentation and/or founder effects influencing population size. There was evidence, however, of an expansion in Ne for R. brelichi at the end of the last glaciation (∼8,500 years ago), which might have contributed to its unexpectedly high heterozygosity. Although the precise factors resulting in high heterozygosity in R. brelichi remain unclear, considering the slow reproductive rate of this species (one litter in 2 years) and the slow absolute nucleotide mutation rate, we surmise that this expansion might have been caused by introgression events, possibly through hybridization with other snub-nosed monkey species or their relatives. More extensive sampling and sequencing of species will afford better resolution of this hypothesis.
Discussion
A comparison of the genome of GSM with the genome of other primates and ruminants has resulted in new phylogenetic and functional insights into primate evolution and genetic adaptations associated with leaf-eating and the consumption of difficult-to-digest resources high in cellulose and hemicellulose. Our analyses have identified the symbiotic microbiota present in the stomach of snub-nosed monkeys, which comprise species capable of metabolizing cellulose. Furthermore, sequencing of the whole genomes of other snub-nosed monkeys enabled a more precise reconstruction of species-specific demographic histories. Although refinement of some of our proposed evolutionary scenarios may require data from functional assays and genomic sequences for Rhinopithecus avunculus (the critically endangered Tonkin snub-nosed monkey that is endemic to Vietnam), such evidence and data are currently unavailable. Nevertheless, the phylogenetic status, evolution of gene families, stomach metagenomics and climate-shaped demographic histories presented here provide a key database and framework for understanding the evolution and functional adaptive patterns of colobines and for developing a program of conservation to promote the survival of these endangered species.
Methods
Sampling and DNA extraction.
The samples used in de novo assembly and resequencing analysis were from the Beijing Wildlife Park (R. roxellana), Beijing Zoo (R. brelichi), Baimaxueshan National Nature Reserve (R. bieti) and Gaoligongshan National Nature Reserve Management Bureau (R. strykeri). The tissue samples used in transcriptome analysis were from a dead female GSM in the Beijing Zoo. Genomic DNA was extracted from whole blood with the help of the Gentra PureGene Blood kit (Qiagen) according to the manufacturer's instructions (Supplementary Note).
Genomic sequencing, assembly and annotation.
Multiple paired-end and mate-pair libraries were constructed with a spanning size range of 180 bp to 20 kb (41 lanes; Supplementary Table 1). All libraries were sequenced on the Illumina HiSeq 2000 platform. After filtering out the adaptor sequences, low-quality reads and duplicate reads, a total of 439.3 Gb of data were retained for assembly. A detailed description of genome size estimation, assembly, annotation, gene structure prediction (based on homology, de novo prediction and RNA-seq data) and functional annotation, and RNA-seq and assembly is included in the Supplementary Note. The draft assembly was evaluated using transcriptome data (Supplementary Table 7), comparing the distributions of GC content across the whole genomes of primates (Supplementary Fig. 5), as well as by mapping the high-quality reads from paired-end libraries with short insert size to the scaffolds using the Burrows-Wheeler Aligner (BWA)41.
Gene family and phylogenetic tree construction.
Gene families were constructed using a hierarchical clustering algorithm (hcluster_sg) and TreeFam42. The phylogenetic tree was reconstructed using the 3,911 single-copy genes shared by GSM and 12 other mammals (human, chimpanzee, gorilla, orangutan, rhesus macaque, crab-eating macaque, gibbon, bushbaby, marmoset, tarsier, mouse lemur and tree shrew) using the maximum-likelihood algorithm as implemented in RAxML software43,44. Protein sequences for these single-copy genes were aligned by MUSCLE45, and protein sequence alignments were transformed back to coding sequence alignments. The divergence times for the taxa analyzed were estimated by Reltime46 on the basis of fourfold-degenerate codon sites. Details for the phylogenetic method and calibration times and a short discussion of the phylogenetic results are available in the Supplementary Note.
Identification of recent transposable elements and ancestor genome reconstruction.
We cataloged recent insertion events for transposable elements (TEs) in four primate genomes. TE annotation for the human, chimpanzee, macaque and GSM genomes was conducted using RepeatMasker (see URLs). We reconstructed the karyotype of the common ancestor of human, macaque and GSM, with tarsier (Tarsius syrichta) and marmoset (Callithrix jacchus) as outgroups, using the genome reconstruction algorithm CARs6. To inspect intervals around breakpoints and identify properties that might help explain why breaks occurred at some positions but not others, we chose 50-kb intervals centered on the ends of conserved segments where the adjacent segments in the ancestor and target species differed.
Positively selected genes.
We used branch-site models47 and likelihood ratio tests (LRTs) in the PAML10 software package to detect PSGs in the GSM genome. P values were computed using the χ2 statistic and corrected for multiple testing by the false discovery rate (FDR) method. ERGs (with significantly higher dN/dS values (ratios of nonsynonymous to synonymous substitutions)) in both the GSM and cattle genomes were identified using the branch model48. The number of sites at which parallel evolution occurred in the GSM and cattle genomes was compared, with the observed number for each genome estimated by comparing present-day sequences to ancestral sequences inferred by the Bayesian method using ANCESTOR49.
Olfactory receptors.
Functional human olfactory receptor gene sequences were downloaded from CRDB (a database of chemosensory receptor gene families in vertebrates)50. The protein sequences for these functional olfactory receptor genes were used in BLAST (TBLASTN) comparisons against the GSM genome with an E-value cutoff of 1 × 10−20. If one genomic locus corresponded to several olfactory receptor query sequences, only the query sequences with the lowest E value were retained. Nonredundant BLAST alignment results were extended in both the 5′ and 3′ directions by 5 kb, and putative olfactory receptor genes in GSM were then predicted using the GeneWise program51. Olfactory receptor genes that contained premature stop codons or frameshift mutations were considered to be pseudogenes. Of the remaining genes, those that encoded proteins of less than 250 amino acids or lacked a complete ORF were classified as truncated genes. The transmembrane regions of the encoded olfactory receptor proteins were assigned according to Man et al.52. All sequences with a gap of 5 amino acids in transmembrane regions were also considered to be pseudogenes. Finally, we reconstructed a phylogenetic tree of the remaining olfactory receptor sequences using the neighbor-joining method implemented in MEGA53.
Additionally, a total of 33 olfactory receptor genes were identified as ERGs in the present study, and the potential target specificity of these olfactory receptor genes in odor perception was determined by comparing the amino acid sequences of the translated GSM olfactory receptor genes to those of human and mouse receptors with previously described information54 (Supplementary Tables 28 and 29).
Functional experiments with the RNASE1 genes.
The ORFs encoding the mature RNASE1 and RNASE1B polypeptides (384 bp) were PCR amplified from the genomic DNA of R. roxellana, Presbytis melalophos and Trachypithecus francoisi and subcloned into the bacterial expression vector pFLAG CTS (Sigma-Aldrich). Experimental procedures for the recombinant protein preparations, including protein isolation, purification and quantification, followed those described in ref. 55. We measured the RNase activity of the recombinant proteins at various pH levels at 25 °C. To evaluate tissue-specific differences in the expression levels of RNASE1 and RNASE1B, multiplex RT-PCR was performed. Each reaction contained 12.5 μl of Premix Ex Taq and 2 μl of cDNA in a total volume of 25 μl. RT-PCR was performed with an initial incubation at 95 °C for 1 min followed by 40 cycles of 5 s at 95 °C and 30 s at 62 °C. The RT-PCR efficiency was calculated for each gene in the multiplex reaction using the formula E = 10−1/slope, with 'slope' being the slope of the five-point standard curve with serial twofold dilutions. The expression of RNASE1 or RNASE1B relative to that for the ACTB gene was calculated using the formula 2−ΔCt, where ΔCt = (Ct RNASE − Ct ACTB). The expression level of the ACTB gene was selected as the calibrator to represent 100% for normalization in each tissue comparison.
Further details on functional experiment appear in the Supplementary Note.
Metagenomics.
DNA was sheared using the Covaris S220 System (Applied Biosystems), and libraries were prepared following a standard protocol from Illumina. The processed sequences were assembled with SOAPdenovo4 using the following parameters: '--K 47 --R --M 3 --d 1'. ORFs were called with the help of the ab initio gene finder MetageneMark56. We used BLASTP to query the protein sequences encoded by the predicted genes in eggNOG57 and iPath58 with an E-value cutoff of <1 × 10−5. PCR amplification was conducted with the 515f/806r primer set that amplifies the V4 region of the 16S rRNA gene. Sequencing was conducted on the Illumina MiSeq platform, and paired-end reads from the original DNA fragments were merged using FLASH59. Sequences were analyzed with the QIIME package60 (Quantitative Insights Into Microbial Ecology) and the UPARSE pipeline61. A description of the annotation of glycoside hydrolases is included in the Supplementary Note.
SNP calling and demographic history reconstruction.
Reads from three resequenced snub-nosed monkeys were aligned to the reference GSM genome using BWA41. Each allele genotype was estimated by mpileup in SAMtools62. Only the mapping reads without gaps and with fewer than five mismatches were included in the identification of SNPs. We identified the ROH for four individuals using the runs of homozygosity tool in PLINK (v.1.07)63 with adjusted parameters (--homozyg-density 500, --homozyg-window-het 1, --homozyg-kb 1000, --homozyg-window-snp 100). To estimate the possibility that functional changes for proteins in Rhinopithecus differed from those in humans, we evaluated the likely effect of a mutation in humans relative to the Rhinopithecus allele as either neutral or deleterious using the EvoD prediction method64, PolyPhen-2 (ref. 65) and SIFT66. Mutations were also evaluated using a prediction model on the basis of consensus between the three methods, which can be found using Bioinformatics Analysis and Research Nexus (BARN) software (see URLs). Mutations were also annotated with their disease status on the basis of HGMD (see URLs), where applicable. The recently developed CoalHMM32,33, which assigns the presence of different genealogies along large multiple alignments, was used to estimate regions of ILS in the genome of snub-nosed monkeys. Inferred historical population sizes were obtained using a PSMC model34. The parameters of PSMC were set to -N30 -t15 -r5 -p “4+25*2+4+6”. The estimated time to the most recent common ancestor (TMRCA) was in units of 2N0 (the estimated effective population size) time, and the mean generation time was set at 10 years for GSM. The parameter μ (substitution rate in nucleotides per year) was estimated under the local clock models, using baseml within the PAML pacakges10.
URLs.
Human Gene Mutation Database (October 2006 release), http://www.hgmd.org/; RepeatMasker, http://www.repeatmasker.org/; Bioinformatics Analysis and Research Nexus (BARN) software, http://barn.asu.edu/software.html.
Accession codes.
The GSM (Rhinopithecus roxellana) whole-genome shotgun project has been deposited at the DNA Data Bank of Japan (DDBJ), the European Molecular Biology Laboratory (EMBL) and GenBank under the accession JABR00000000. The version described in this paper is the first version, JABR01000000. The BioProject accession number for the snub-nosed monkey genome sequence is PRJNA230020. Assembly-based SNPs and SNPs derived from short-read sequence data have been deposited in dbSNP. All short-read data have been deposited in the Sequence Read Archive (SRP033389). Snub-nosed monkey RNA-seq data have been deposited in the Gene Expression Omnibus under accession GSE53597.
Accession codes
Primary accessions
BioProject
DDBJ/GenBank/EMBL
Gene Expression Omnibus
Sequence Read Archive
References
Milton, K. Food and Evolution: Toward a Theory of Human Food Habits (eds. Harris M. & Ross, E.B.) 93–116 (Temple University Press, Philadelphia, 1987).
Kay, R.N.B. & Davies, A.G. Colobine Monkeys: Their Ecology, Behaviour and Evolution (eds. Davies, A.G. & Oates, J.F.) 229–250 (Cambridge University Press, Cambridge, UK, 1994).
Li, B.G., Pan, R.L. & Oxnard, C.E. Extinction of snub-nosed monkeys in China during the past 400 years. Int. J. Primatol. 23, 1227–1244 (2002).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Deininger, P. Alu elements: know the SINEs. Genome Biol. 12, 236 (2011).
Ma, J. et al. Reconstructing contiguous regions of an ancestral genome. Genome Res. 16, 1557–1565 (2006).
Esnault, C., Maestre, J. & Heidmann, T. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 24, 363–367 (2000).
Dickinson, D.P., Thiesse, M. & Hicks, M.J. Expression of type 2 cystatin genes CST1–CST5 in adult human tissues and the developing submandibular gland. DNA Cell Biol. 21, 47–65 (2002).
Mau, M. et al. First identification of tannin-binding proteins in saliva of Papio hamadryas using MS/MS mass spectrometry. Am. J. Primatol. 73, 896–902 (2011).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Milton, K. Physiological ecology of howlers (Alouatta): energetic and digestive considerations and comparison with the Colobinae. Int. J. Primatol. 19, 513–548 (1998).
Matsubara, Y. et al. Molecular cloning of cDNAs encoding rat and human medium-chain acyl-CoA dehydrogenase and assignment of the gene to human chromosome 1. Proc. Natl. Acad. Sci. USA 83, 6543–6547 (1986).
Uchida, Y., Izai, K., Orii, T. & Hashimoto, T. Novel fatty acid β-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J. Biol. Chem. 267, 1034–1041 (1992).
Sillén, A. et al. Spectrum of mutations and sequence variants in the FALDH gene in patients with Sjogren-Larsson syndrome. Hum. Mutat. 12, 377–384 (1998).
Gilad, Y., Wiebe, V., Przeworski, M., Lancet, D. & Paabo, S. Loss of olfactory receptor genes coincides with the acquisition of full trichromatic vision in primates. PLoS Biol. 2, e5 (2004).
Niimura, Y. & Nei, M. Extensive gains and losses of olfactory receptor genes in mammalian evolution. PLoS ONE 2, e708 (2007).
Zhang, J., Zhang, Y.P. & Rosenberg, H.F. Adaptive evolution of a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nat. Genet. 30, 411–415 (2002).
Zhang, J. Parallel adaptive origins of digestive RNases in Asian and African leaf monkeys. Nat. Genet. 38, 819–823 (2006).
Schienman, J.E. Duplication and divergence of 2 distinct pancreatic ribonuclease genes in leaf-eating African and Asian colobine monkeys. Mol. Biol. Evol. 23, 1465–1479 (2006).
Yu, L. et al. Adaptive evolution of digestive RNASE1 genes in leaf-eating monkeys revisited: new insights from ten additional Colobines. Mol. Biol. Evol. 27, 121–131 (2010).
von Rosenvinge, E.C. et al. Immune status, antibiotic medication and pH are associated with changes in the stomach fluid microbiota. ISME J. 7, 1354–1366 (2013).
Zhu, L., Wu, Q., Dai, J., Zhang, S. & Wei, F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc. Natl. Acad. Sci. USA 108, 17714–17719 (2011).
Pope, P.B. et al. Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc. Natl. Acad. Sci. USA 107, 14793–14798 (2010).
Warnecke, F. et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565 (2007).
Liedigk, R. et al. Evolutionary history of the odd-nosed monkeys and the phylogenetic position of the newly described Myanmar snub-nosed monkey Rhinopithecus strykeri. PLoS ONE 7, e37418 (2012).
Prado-Martinez, J. et al. Great ape genetic diversity and population history. Nature 499, 471–475 (2013).
Scally, A. et al. Insights into hominid evolution from the gorilla genome sequence. Nature 483, 169–175 (2012).
Quan, G.Q. & Xie,, J.Y. Research on the Golden Monkey (Science and Education Publishing House, Beijing, 2002).
Xiang, Z.F. et al. Current status and conservation of the gray snub-nosed monkey Rhinopithecus brelichi (Colobinae) in Guizhou, China. Biol. Conserv. 142, 469–476 (2009).
Kumar, S. et al. Phylomedicine: an evolutionary telescope to explore and diagnose the universe of disease mutations. Trends Genet. 27, 377–386 (2011).
Zheng, B., Xu, Q. & Shen, Y. The relationship between climate change and Quaternary glacial cycles on the Qinghai-Tibetan Plateau: review and speculation. Quat. Int. 97-98, 93–101 (2002).
Hobolth, A., Christensen, O.F., Mailund, T. & Schierup, M.H. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 3, e7 (2007).
Mailund, T., Dutheil, J.Y., Hobolth, A., Lunter, G. & Schierup, M.H. Estimating divergence time and ancestral effective population size of Bornean and Sumatran orangutan subspecies using a coalescent hidden Markov model. PLoS Genet. 7, e1001319 (2011).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Zhao, S. et al. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat. Genet. 45, 67–71 (2013).
Lisiecki, L.E. & Raymo, M.E.A. Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanography 20, PA103 (2005).
Miller, W. et al. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc. Natl. Acad. Sci. USA 109, E2382–E2390 (2012).
Bintanja, R. & van de Wal, R.S. North American ice-sheet dynamics and the onset of 100,000-year glacial cycles. Nature 454, 869–872 (2008).
Sun, Y.B. & An, Z.S. Late Pliocene-Pleistocene changes in mass accumulation rates of eolian deposits on the central Chinese Loess Plateau. J. Geophys. Res. 110, D23101 (2005).
Li, T. et al. High-resolution climate variability of southwest China during 57–70 ka reflected in a stalagmite δ18O record from Xinya Cave. Sci. China Ser. D Earth Sci. 50, 1202–1208 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 34, D572–D580 (2006).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood–based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771 (2008).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Tamura, K. et al. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 109, 19333–19338 (2012).
Zhang, J., Nielsen, R. & Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479 (2005).
Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573 (1998).
Zhang, J. & Nei, M. Accuracies of ancestral amino acid sequences inferred by the parsimony, likelihood, and distance methods. J. Mol. Evol. 44, S139–S146 (1997).
Dong, D., Jin, K., Wu, X. & Zhong, Y. CRDB: database of chemosensory receptor gene families in vertebrate. PLoS ONE 7, e31540 (2012).
Birney, E., Clamp, M. & Durbin, R. Genewise and genomewise. Genome Res. 14, 988–995 (2004).
Man, O., Gilad, Y. & Lancet, D. Prediction of the odorant binding site of olfactory receptor proteins by human-mouse comparisons. Protein Sci. 13, 240–254 (2004).
Kumar, S., Nei, M., Dudley, J. & Tamura, K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 9, 299–306 (2008).
Nguyen, D.T. et al. The complete swine olfactory subgenome: expansion of the olfactory gene repertoire in the pig genome. BMC Genomics 13, 584 (2012).
Rosenberg, H.F. & Dyer, K.D. Eosinophil cationic protein and eosinophil-derived neurotoxin. J. Biol. Chem. 270, 21539–21544 (1995).
Zhu, W., Lomsadze, A. & Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38, e132 (2010).
Muller, J. et al. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 38, D190–D195 (2010).
Letunic, I., Yamada, T., Kanehisa, M. & Bork, P. iPath: Interactive exploration of biochemical pathways and networks. Trends Biochem. Sci. 33, 101–103 (2008).
Magocč, T. & Salzberg, S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Caporaso, J.G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Edgar, R.C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Kumar, S., Sanderford, M., Gray, V.E., Ye, J. & Liu, L. Evolutionary diagnosis method for variants in personal exomes. Nat. Methods 9, 855–856 (2012).
Adzhubei, I.A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Ng, P.C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Acknowledgements
This project was supported by grants from the National Key Technology R&D Program of China (2013BAD03B02), the Natural Science Foundation of China (31270420, 41371071, 31372185 and 31130061), the Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-Q-7-2) and the US National Institutes of Health (HG002096-12).
Author information
Authors and Affiliations
Contributions
Ming Li conceived the study and designed the project. X.Z., B.W. and J.Z. managed the project. B.W. prepared samples and implemented the point mutation experiment. X.Z. coordinated genome assembly, annotation and bioinformatics analysis. Q.P. and J.Z. performed genome assembly and annotation. X.Z., S.K., Q.P., X.S., W.J., Y.L., W.Z., G.L. and X.M. performed genetic analyses. X.Z., B.W. and Q.P. discussed the data. X.Z. and B.W. wrote the manuscript with contributions from Q.P., S.K., Mingzhou Li, P.A.G., M.W.B., R.L., Z.L., H.P., B.R., H.R., C.C., D.W., F.S., Y.H., Y.T., C.Z., P.Z., Z.X., J.C., H.W., Z.C., Z.J., B.L., G.Y. and C.R. All authors contributed to data interpretation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–28, Supplementary Tables 1–41 and Supplementary Note. (PDF 7092 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Zhou, X., Wang, B., Pan, Q. et al. Whole-genome sequencing of the snub-nosed monkey provides insights into folivory and evolutionary history. Nat Genet 46, 1303–1310 (2014). https://doi.org/10.1038/ng.3137
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3137
This article is cited by
-
Whole-genome resequencing of the native sheep provides insights into the microevolution and identifies genes associated with reproduction traits
BMC Genomics (2023)
-
Comparative study of the gut microbiota in three captive Rhinopithecus species
BMC Genomics (2023)
-
Current advances in primate genomics: novel approaches for understanding evolution and disease
Nature Reviews Genetics (2023)
-
Birth-and-death evolution of ribonuclease 9 genes in Cetartiodactyla
Science China Life Sciences (2023)
-
Insights into cold tolerance in sable (Martes zibellina) from the adaptive evolution of lipid metabolism
Mammalian Biology (2021)
