
Abstract
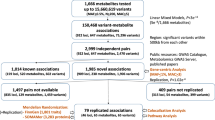
Genome-wide association scans with high-throughput metabolic profiling provide unprecedented insights into how genetic variation influences metabolism and complex disease. Here we report the most comprehensive exploration of genetic loci influencing human metabolism thus far, comprising 7,824 adult individuals from 2 European population studies. We report genome-wide significant associations at 145 metabolic loci and their biochemical connectivity with more than 400 metabolites in human blood. We extensively characterize the resulting in vivo blueprint of metabolism in human blood by integrating it with information on gene expression, heritability and overlap with known loci for complex disorders, inborn errors of metabolism and pharmacological targets. We further developed a database and web-based resources for data mining and results visualization. Our findings provide new insights into the role of inherited variation in blood metabolic diversity and identify potential new opportunities for drug development and for understanding disease.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Garrod, A.E. Inborn Factors in Disease (Oxford University Press, Oxford, 1931).
Kettunen, J. et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat. Genet. 44, 269–276 (2012).
Sabatine, M.S. et al. Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation 112, 3868–3875 (2005).
Holmes, E. et al. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 453, 396–400 (2008).
Sreekumar, A. et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457, 910–914 (2009).
Bictash, M. et al. Opening up the “Black Box”: metabolic phenotyping and metabolome-wide association studies in epidemiology. J. Clin. Epidemiol. 63, 970–979 (2010).
Backshall, A., Sharma, R., Clarke, S.J. & Keun, H.C. Pharmacometabonomic profiling as a predictor of toxicity in patients with inoperable colorectal cancer treated with capecitabine. Clin. Cancer Res. 17, 3019–3028 (2011).
Wang, T.J. et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 17, 448–453 (2011).
Suhre, K. & Gieger, C. Genetic variation in metabolic phenotypes: study designs and applications. Nat. Rev. Genet. 13, 759–769 (2012).
Gieger, C. et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet. 4, e1000282 (2008).
Suhre, K. et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature 477, 54–60 (2011).
Nicholson, G. et al. Human metabolic profiles are stably controlled by genetic and environmental variation. Mol. Syst. Biol. 7, 525 (2011).
Sanseau, P. et al. Use of genome-wide association studies for drug repositioning. Nat. Biotechnol. 30, 317–320 (2012).
Krumsiek, J. et al. Mining the unknown: a systems approach to metabolite identification combining genetic and metabolic information. PLoS Genet. 8, e1003005 (2012).
Lawlor, D.A., Harbord, R.M., Sterne, J.A., Timpson, N. & Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012).
Frazer, K.A. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007).
Pe'er, I., Yelensky, R., Altshuler, D. & Daly, M.J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 32, 381–385 (2008).
Rhee, E.P. et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab. 18, 130–143 (2013).
Illig, T. et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 42, 137–141 (2010).
Suhre, K. et al. A genome-wide association study of metabolic traits in human urine. Nat. Genet. 43, 565–569 (2011).
Nicholson, G. et al. A genome-wide metabolic QTL analysis in Europeans implicates two loci shaped by recent positive selection. PLoS Genet. 7, e1002270 (2011).
Shrawder, E. & Martinez-Carrion, M. Evidence of phenylalanine transaminase activity in the isoenzymes of aspartate transaminase. J. Biol. Chem. 247, 2486–2492 (1972).
Lee, H.C. et al. Caenorhabditis elegans mboa-7, a member of the MBOAT family, is required for selective incorporation of polyunsaturated fatty acids into phosphatidylinositol. Mol. Biol. Cell 19, 1174–1184 (2008).
Hu, C.A. et al. Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol. Cell. Biochem. 295, 85–92 (2007).
Krumsiek, J., Suhre, K., Illig, T., Adamski, J. & Theis, F.J. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst. Biol. 5, 21 (2011).
Schadt, E.E. et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 6, e107 (2008).
Grundberg, E. et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat. Genet. 44, 1084–1089 (2012).
Kim, D.K. et al. The human T-type amino acid transporter-1: characterization, gene organization, and chromosomal location. Genomics 79, 95–103 (2002).
Kaper, T. et al. Nanosensor detection of an immunoregulatory tryptophan influx/kynurenine efflux cycle. PLoS Biol. 5, e257 (2007).
Mootha, V.K. & Hirschhorn, J.N. Inborn variation in metabolism. Nat. Genet. 42, 97–98 (2010).
Köttgen, A. et al. New loci associated with kidney function and chronic kidney disease. Nat. Genet. 42, 376–384 (2010).
Xie, W. et al. Genetic variants associated with glycine metabolism and their role in insulin sensitivity and type 2 diabetes. Diabetes 62, 2141–2150 (2013).
Kikuchi, G., Motokawa, Y., Yoshida, T. & Hiraga, K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 84, 246–263 (2008).
Moayyeri, A., Hammond, C.J., Hart, D.J. & Spector, T.D. The UK adult twin registry (TwinsUK Resource). Twin Res. Hum. Genet. 16, 144–149 (2013).
Wichmann, H.E., Gieger, C. & Illig, T. KORA-gen—resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen 67 (suppl. 1), S26–S30 (2005).
Abecasis, G.R., Cherny, S.S., Cookson, W.O. & Cardon, L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002).
Kutalik, Z., Whittaker, J., Waterworth, D., Beckmann, J.S. & Bergmann, S. Novel method to estimate the phenotypic variation explained by genome-wide association studies reveals large fraction of the missing heritability. Genet. Epidemiol. 35, 341–349 (2011).
Willer, C.J., Li, Y. & Abecasis, G.R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Boker, S. et al. OpenMx: an open source extended structural equation modeling framework. Psychometrika 76, 306–317 (2011).
1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Zuk, O., Hechter, E., Sunyaev, S.R. & Lander, E.S. The mystery of missing heritability: genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 109, 1193–1198 (2012).
Hao, T., Ma, H.W., Zhao, X.M. & Goryanin, I. Compartmentalization of the Edinburgh human metabolic network. BMC Bioinformatics 11, 393 (2010).
Schomburg, I. et al. BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res. 41, D764–D772 (2013).
Smith, G.D. & Ebrahim, S. 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 32, 1–22 (2003).
Acknowledgements
For TwinsUK, we thank the Genotyping Facilities at the Wellcome Trust Sanger Institute and the Center for Inherited Disease Research (CIDR)/US National Institutes of Health (NIH) for SNP genotyping. The KORA Study Group consists of A. Peters (speaker), J. Heinrich, R. Holle, R. Leidl, C. Meisinger, K. Strauch and their coworkers, who are responsible for the design and implementation of the KORA studies. For KORA, we thank P. Lichtner, G. Eckstein, G. Fischer, T. Strom, the Helmholtz Zentrum München genotyping staff and the field staff of the MONICA/KORA Augsburg studies. We also thank G. Fischer (KORA) and G. Surdulescu (TwinsUK) for sample handling and H. Chavez (KORA) and D. Hodgkiss (TwinsUK) for sample shipment. We are grateful to the MuTHER investigators for the transcriptomic data. Finally, we wish to express our appreciation to all study participants of the TwinsUK and KORA studies for donating their blood and time.
Part of this work was funded by Pfizer Worldwide Research and Development. For TwinsUK, the study was funded by the Wellcome Trust; European Community's Seventh Framework Programme (FP7/2007-2013). The study also receives support from the National Institute for Health Research (NIHR) BioResource Clinical Research Facility and Biomedical Research Centre based at Guy's and St Thomas' National Health Service (NHS) Foundation Trust and King's College London. T.D.S. is the holder of a European Research Council (ERC) Advanced Principal Investigator award. SNP genotyping was performed by the Wellcome Trust Sanger Institute and the National Eye Institute via NIH/CIDR. The KORA (Kooperative Gesundheitsforschung in der Region Augsburg) research platform and the MONICA Augsburg studies were initiated and financed by the Helmholtz Zentrum München National Research Center for Environmental Health, which is funded by the German Federal Ministry of Education, Science, Research and Technology and by the state of Bavaria. This study was supported by a grant from the German Federal Ministry of Education and Research (BMBF) to the German Center for Diabetes Research (DZD). The German National Genome Research Network financed part of this work (NGFNPlus 01GS0823). Computing resources have been made available by the Leibniz Supercomputing Centre of the Bavarian Academy of Sciences and Humanities (HLRB project h1231) and by the DEISA Extreme Computing Initiative (project PHAGEDA). Part of this research was supported within the Munich Center of Health Sciences (MC Health) as part of LMUinnovativ. S.-Y.S. is supported by a Post-Doctoral Research Fellowship from the Oak Foundation. F.J.T. is supported by an ERC starting grant (LatentCauses). J.K. is supported by the German Research Foundation (SPP 1395, InKoMBio) and by a grant from the German Helmholtz Postdoctoral Programme. K.S. is supported by Biomedical Research Program funds at Weill Cornell Medical College in Qatar, a program funded by the Qatar Foundation. C.G. is supported by the European Union's Seventh Framework project MIMOmics (FP7-Health-F5-2012-305280) and by the Russian Foundation for Basic Research (RFBR)-Helmholtz research group program. N.S. is supported by the Wellcome Trust (grants WT098051 and WT091310) and by the European Commission (EUFP7 EPIGENESYS grant 257082 and BLUEPRINT grant HEALTH-F5-2011-282510). J.B.R. and V.F. are supported by the Canadian Institutes of Health Research, Fonds du Recherche du Science Québec and the Québec Consortium for Drug Discovery.
The Pfizer colleagues dedicate this manuscript to the memory of our friend Phoebe Roberts, whose passion for text mining, molecular biology and drug discovery contributed to the identification of causal genes in this research and to our work in general.
Author information
Authors and Affiliations
Consortia
Contributions
Study organization: M.J.B., K.S., C.G., G.K. and N.S. Manuscript preparation: S.-Y.S., J.K., K.S., G.K. and N.S. Data collection: R.P.M. and M.V.M. Analysis of associations: S.-Y.S., A.-K.P., J.H., G.K. and N.S. Locus bioinformatics annotation: E.B.F., D.Z., R.S., V.F., L.C., L.J.V., K.W., V.W., P.R., L.X., J.B.R., J.P.O. and G.K. GGM network: J.K., F.J.T. and G.K. Supplementary websites and online resources: M.A., G.K. and N.S. Provision of materials, data and analysis tools: T.D.S., K.S., G.K., E.B.F., M.W., C.G., J.T., I.E., A.M.V., C.L.H., T.-P.Y., C.M., S.L.J. and E.G.
Corresponding authors
Ethics declarations
Competing interests
M.V.M. and R.P.M. are employees of Metabolon, Inc. E.B.F., C.L.H., V.W., D.Z., P.R., L.X., S.L.J., J.T. and M.J.B. are full-time employees and shareholders of Pfizer.
Additional information
A full list of members and affiliations appears in the Supplementary Note.
Integrated supplementary information
Supplementary Figure 2 Manhattan plots.
Association results for raw metabolite concentrations are shown for genome-wide SNPs. Top, TwinsUK; bottom, KORA. Only SNPs with P < 1 ×10–6 are displayed. The green line indicates the genome-wide cutoff of P < 1.03 ×10–10. Loci with P value < 1 ×10–30 are indicated with a red symbol.
Supplementary Figure 3 Comparison of imputation based on HapMap 2 and 1000 Genomes Project data.
Correlation between (a) minor allele frequency (MAF) in meta-analysis, (b) association P value in meta-analysis (on a –log10 scale) and (c) average variance explained for the most significant SNPs selected from imputation based on either the HapMap 2 (x axis) or 1000 Genomes Project (y axis) panels. High correlations between the HapMap 2 and 1000 Genomes Project data sets support the view that metabolic associations are driven by common variants that are well tagged by HapMap 2 imputation. (d,e) One exception is the CYP3A4-CYP3A5-CYP3A7 locus where the 1000 Genomes Project scan reveals an additional variant (rs10278040) with greater association and variance explained for androsterone sulfate than for the corresponding HapMap 2 variant (rs148982377). (HapMap 2: rs148982377, MAF = 0.038, P = 7.65 ×10–244, R2 = 15.6%; 1000 Genomes Project: rs10278040, MAF = 0.042, P = 8.82 ×10–113, = 10.3%.)
= 10.3%.)
Supplementary Figure 4 Interaction between NAT8 and PYROXD2 variants.
(a) Box plots of PYROXD2 and NAT8 transcript levels in fat, skin and LCLs as a function of the genotype conformation between the two variants rs10469966 (NAT8) and rs4488133 (PYROXD2). (b) Summary of association and interaction effects at the two loci, summarizing association statistics and variances explained under the single-SNP, additive and interaction models. An ANOVA F test was used to test the significance of the interactive model over the additive model. The association test with X-12093 reported here is based on a combined TwinsUK and KORA data set; all other analyses were carried out using unrelated TwinsUK singletons. See also Figure 4.
Supplementary Figure 5 Cardiovascular disease and hypertension metabolic subnetwork.
Network data were annotated with expert knowledge to illustrate correlations between molecular relationships and knowledge on blood pressure regulation, blood coagulation and known molecular risk factors for cardiovascular disease and hypertension. Black nodes and edges represent a subnetwork from the metabolite network in Figure 2. This subnetwork contains metabolites (circular nodes) and genes (diamond-shaped nodes) of the fibrinogen cleavage (left) and kininogen/kinin (right) systems and their interconnections as derived from our data. Gray nodes and edges, annotations of biochemical function based on expert knowledge27; colored nodes and edges, reported associations based on genome-wide studies for blood pressure regulation (orange), blood coagulation (blue) and cholesterol levels (purple; information in Supplementary Table 6).
Supplementary information
Supplementary Text and Figures
Supplementary Note and Supplementary Figures 1–5 (PDF 4947 kb)
Supplementary Tables 1–14
Supplementary Tables 1–14 (XLSX 12504 kb)
Rights and permissions
About this article

Cite this article
Shin, SY., Fauman, E., Petersen, AK. et al. An atlas of genetic influences on human blood metabolites. Nat Genet 46, 543–550 (2014). https://doi.org/10.1038/ng.2982
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2982
This article is cited by
-
Causal association between 637 human metabolites and ovarian cancer: a mendelian randomization study
BMC Genomics (2024)
-
Mendelian randomization analyses of known and suspected risk factors and biomarkers for myasthenia gravis overall and by subtypes
BMC Neurology (2024)
-
The impact of lipidome on breast cancer: a Mendelian randomization study
Lipids in Health and Disease (2024)
-
Causal associations between gut microbiota, metabolites and asthma: a two-sample Mendelian randomization study
BMC Pulmonary Medicine (2024)
-
Causal relationships of metabolites with allergic diseases: a trans-ethnic Mendelian randomization study
Respiratory Research (2024)
