
Abstract
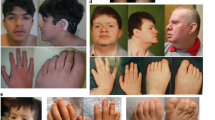
Cantú syndrome is characterized by congenital hypertrichosis, distinctive facial features, osteochondrodysplasia and cardiac defects. By using family-based exome sequencing, we identified a de novo mutation in ABCC9. Subsequently, we discovered novel dominant missense mutations in ABCC9 in 14 of the 16 individuals with Cantú syndrome examined. The ABCC9 protein is part of an ATP-dependent potassium (KATP) channel that couples the metabolic state of a cell with its electrical activity. All mutations altered amino acids in or close to the transmembrane domains of ABCC9. Using electrophysiological measurements, we show that mutations in ABCC9 reduce the ATP-mediated potassium channel inhibition, resulting in channel opening. Moreover, similarities between the phenotype of individuals with Cantú syndrome and side effects from the KATP channel agonist minoxidil indicate that the mutations in ABCC9 result in channel opening. Given the availability of ABCC9 antagonists, our findings may have direct implications for the treatment of individuals with Cantú syndrome.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


Change history
03 June 2012
In the version of this article initially published online, author David J. Amor's name was given incorrectly as David J.D. Amor. The error has been corrected for the print, PDF and HTML versions of this article.
References
Cantú, J.M., Garcia-Cruz, D., Sanchez-Corona, J., Hernandez, A. & Nazara, Z. A distinct osteochondrodysplasia with hypertrichosis—individualization of a probable autosomal recessive entity. Hum. Genet. 60, 36–41 (1982).
Garcia-Cruz, D. et al. Congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly: further delineation of a new genetic syndrome. Am. J. Med. Genet. 69, 138–151 (1997).
Nevin, N.C., Mulholland, H.C. & Thomas, P.S. Congenital hypertrichosis, cardiomegaly and mild osteochondrodysplasia. Am. J. Med. Genet. 66, 33–38 (1996).
Rosser, E.M. et al. Three patients with the osteochondrodysplasia and hypertrichosis syndrome—Cantú syndrome. Clin. Dysmorphol. 7, 79–85 (1998).
Robertson, S.P. et al. Congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly: Cantú syndrome. Am. J. Med. Genet. 85, 395–402 (1999).
Concolino, D., Formicola, S., Camera, G. & Strisciuglio, P. Congenital hypertrichosis, cardiomegaly, and osteochondrodysplasia (Cantú syndrome): a new case with unusual radiological findings. Am. J. Med. Genet. 92, 191–194 (2000).
Lazalde, B., Sanchez-Urbina, R., Nuno-Arana, I., Bitar, W.E. & de Lourdes Ramirez-Duenas, M. Autosomal dominant inheritance in Cantú syndrome (congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly). Am. J. Med. Genet. 94, 421–427 (2000).
Engels, H. et al. Further case of Cantú syndrome: exclusion of cryptic subtelomeric chromosome aberrations. Am. J. Med. Genet. 111, 205–209 (2002).
Grange, D.K., Lorch, S.M., Cole, P.L. & Singh, G.K. Cantú syndrome in a woman and her two daughters: further confirmation of autosomal dominant inheritance and review of the cardiac manifestations. Am. J. Med. Genet. A. 140, 1673–1680 (2006).
Tan, T.Y. et al. A patient with monosomy 1p36, atypical features and phenotypic similarities with Cantú syndrome. Am. J. Med. Genet. A. 139, 216–220 (2005).
Scurr, I. et al. Cantú syndrome: report of nine new cases and expansion of the clinical phenotype. Am. J. Med. Genet. A. 155A, 508–518 (2011).
Graziadio, C. et al. Short-term follow-up of a Brazilian patient with Cantú syndrome. Am. J. Med. Genet. A. 155A, 1184–1188 (2011).
Babenko, A.P. & Bryan, J. SUR domains that associate with and gate KATP pores define a novel gatekeeper. J. Biol. Chem. 278, 41577–41580 (2003).
Shi, N.Q., Ye, B. & Makielski, J.C. Function and distribution of the SUR isoforms and splice variants. J. Mol. Cell. Cardiol. 39, 51–60 (2005).
Shorter, K., Farjo, N.P., Picksley, S.M. & Randall, V.A. Human hair follicles contain two forms of ATP-sensitive potassium channels, only one of which is sensitive to minoxidil. FASEB J. 22, 1725–1736 (2008).
Bienengraeber, M. et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 36, 382–387 (2004).
Olson, T.M. et al. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat. Clin. Pract. Cardiovasc. Med. 4, 110–116 (2007).
Flagg, T.P., Enkvetchakul, D., Koster, J.C. & Nichols, C.G. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol. Rev. 90, 799–829 (2010).
Mikhailov, M.V. et al. 3-D structural and functional characterization of the purified KATP channel complex Kir6.2–SUR1. EMBO J. 24, 4166–4175 (2005).
Moreau, C., Prost, A.L., Derand, R. & Vivaudou, M. SUR, ABC proteins targeted by KATP channel openers. J. Mol. Cell. Cardiol. 38, 951–963 (2005).
Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476 (2006).
Schwanstecher, M. et al. Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J. 17, 5529–5535 (1998).
Rafiq, M. et al. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care 31, 204–209 (2008).
Harakalova, M. et al. Multiplexed array-based and in-solution genomic enrichment for flexible and cost-effective targeted next-generation sequencing. Nat. Protoc. 6, 1870–1886 (2011).
Mokry, M. et al. Accurate SNP and mutation detection by targeted custom microarray–based genomic enrichment of short-fragment sequencing libraries. Nucleic Acids Res. 38, e116 (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Nijman, I.J. et al. Mutation discovery by targeted genomic enrichment of multiplexed barcoded samples. Nat. Methods 7, 913–915 (2010).
Ishihara, K. & Ehara, T. Two modes of polyamine block regulating the cardiac inward rectifier K+ current IK1 as revealed by a study of the Kir2.1 channel expressed in a human cell line. J. Physiol. (Lond.) 556, 61–78 (2004).
Acknowledgements
We thank the families of our subjects for participating in this study. We thank J.A. Sánchez-Chapula (Centro Universitario de Investigaciones Biomédicas de la Universidad de Colima) for providing wild-type KCNJ11 and ABCC9 expression constructs and N. Lansu and E. de Bruijn for technical support. F.W.A. is supported by a clinical fellowship from the Netherlands Organisation for Health Research and Development (ZonMw; 90700342). G.v.H. is supported by a Veni Grant from the Netherlands Organisation for Health Research and Development. This study was financially supported by the Child Health Priority Program of the University Medical Center Utrecht.
Author information
Authors and Affiliations
Contributions
J.J.T.v.H., P.A.T., D.J.A., L.C.W., E.P.K., C.L.S.T., D.S., S.G.-M., M.M.L., A.R., F.W.A., H.M.B., M.E.S., I.J.S., S.F.S., J.J.v.d.S. and M.M.v.H. characterized individuals with Cantú syndrome and collected clinical data. M.H., K.D., I.R., W.P.K. and G.v.H. performed genetic studies, and M.H., S.v.L. and I.J.N. performed next-generation sequencing data analysis. H.V. and G.V. performed protein modeling studies. H.T., M.B.R. and M.A.G.v.d.H. designed and performed patch clamp experiments. G.v.H., M.H. and J.J.T.v.H. prepared the final manuscript. M.M.v.H., N.V.K., W.P.K., G.v.H. and E.C. supervised the study. All authors critically contributed to the study design and the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Tables 1–5, Supplementary Figures 1 and 2 and Supplementary Note (PDF 3209 kb)
Rights and permissions
About this article
Cite this article
Harakalova, M., van Harssel, J., Terhal, P. et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat Genet 44, 793–796 (2012). https://doi.org/10.1038/ng.2324
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2324
This article is cited by
-
Lymphedema as first clinical presentation of Cantu Syndrome: reversed phenotyping after identification of gain-of-function variant in ABCC9
European Journal of Human Genetics (2023)
-
The inhibition mechanism of the SUR2A-containing KATP channel by a regulatory helix
Nature Communications (2023)
-
Structural identification of vasodilator binding sites on the SUR2 subunit
Nature Communications (2022)
-
Gene expression profile of the murine ischemic retina and its response to Aflibercept (VEGF-Trap)
Scientific Reports (2021)
-
Clinical genetics in transition—a comparison of genetic services in Estonia, Finland, and the Netherlands
Journal of Community Genetics (2021)
