
Abstract
Self-recognition and self-discrimination within complex mixtures are of fundamental importance in biological systems, which entirely rely on the preprogrammed monomer sequences and homochirality of biological macromolecules. Here we report artificial chirality- and sequence-selective successive self-sorting of chiral dimeric strands bearing carboxylic acid or amidine groups joined by chiral amide linkers with different sequences through homo- and complementary-duplex formations. A mixture of carboxylic acid dimers linked by racemic-1,2-cyclohexane bis-amides with different amide sequences (NHCO or CONH) self-associate to form homoduplexes in a completely sequence-selective way, the structures of which are different from each other depending on the linker amide sequences. The further addition of an enantiopure amide-linked amidine dimer to a mixture of the racemic carboxylic acid dimers resulted in the formation of a single optically pure complementary duplex with a 100% diastereoselectivity and complete sequence specificity stabilized by the amidinium–carboxylate salt bridges, leading to the perfect chirality- and sequence-selective duplex formation.
Similar content being viewed by others

Introduction
High-fidelity self-recognition and self-discrimination are of principal importance in living systems1, which enable biological macromolecules, such as proteins and DNA, to self-organize into uniform quaternary and double helical structures with a controlled handedness, respectively, through noncovalent interactions even within complex mixtures of subunits or molecular strands with a similar shape, size and sequence, thereby providing sophisticated functions that are essential for human life1,2. Such an incredible self-sorting performance observed in biological systems ultimately relies on the preprogrammed monomer sequences and homochirality of their building blocks and components2,3. Recent advances in the total chemical synthesis of unnatural D-proteins4,5 and L-DNA6 clearly revealed the indispensable role of the homochirality along with the monomer sequences of natural L-proteins and D-DNA towards chiral specificities in enantioselective reactions and complementary double-helix formations, respectively.
In organic and supramolecular chemistry, the control of the size and shape7,8,9,10,11,12,13, and topology14,15 as well as the handedness16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31 of supramolecular assemblies from mixtures of a variety of components via spontaneous self-sorting has become one of the urgent and emerging topics10,32,33 because it will not only enrich our understanding of the principles underling the precise recognition behaviour of biological macromolecules but also contribute to the development of novel supramolecular catalysts34, optoelectrical devices35 and sensors36. In most cases32,33, however, self-sorting has been achieved in a size- and shape-7,8,9,10,11,12,13 or topology-selective way14,15,28 through the formation of macrocycles11,15, capsules or cages13,17,22 and helices23,24,27,30,31,37, while a limited number of supramolecular systems undergoes chiral self-sorting, producing either homochiral16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32 or heterochiral assemblies32,38,39,40, which is mostly relevant to the biological processes as well as the state-of-the-art asymmetric catalysis41 and optical resolution on crystallization42.
Here we report artificial chirality- and sequence-selective chiral self-sorting of dimeric strands consisting of carboxylic acid or amidine groups joined by chiral amide linkers with different sequences (NHCO or CONH) through specific homoduplex and subsequent hetero- (complementary-) duplex formations with implications for biological double helices in DNA. DNA-like double helices have a significant advantage in the precise recognition of the monomer sequence14,43 and chain length between the complementary strands44. Although hydrogen-bond-driven peptide nucleic acids45 and metal-coordinate-bonded helicates7,8,9,46 are known to show self-sorting with respect to their sequences and/or chain lengths, chirality- and sequence-selective successive self-sorting is currently unknown.
Results
Design and synthesis of carboxylic acid and amidine dimers
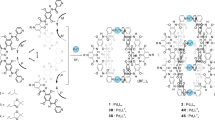
Our molecular design is mainly based on the previously reported heterodouble helices composed of complementary dimer strands with m-terphenyl backbones intertwined through amidinium–carboxylate salt bridges (Fig. 1a, AA·CC)44,47,48,49,50. Owing to the high tolerance of the salt bridges towards various functional groups, a variety of linkers (L) could be introduced while maintaining the double-stranded helical structures with a one-handed helical sense biased by the chirality introduced on the amidine residues49. During the course of our study, we also found that an achiral carboxylic acid dimer joined by a p-diethynylbenzene linker with n-octyl (RA) substituents self-associated into a racemic homodouble helix through interstrand hydrogen bonds between the carboxy groups (Fig. 1a, (CC2))51.

(a) Structures of heterodouble helix (AA·CC) and homodouble helix (CC2). (b) Structures of carboxylic acid and amidine dimer strands linked through chiral or meso amide residues (X=L1 or L2). (c) Schematic representation of chirality- and sequence-selective successive self-sorting via specific homo- and complementary-duplex formations. Carboxylic acid dimers of rac-1a and rac-2a with different linker amide sequences form homo/hetero- (i) and homochiral (ii) duplexes, respectively. A mixture of rac-1a and rac-2a completely self-sorts in sequence-selective way to give duplexes of 1a and 2a (iii). Only one enantiomer among rac-1a and rac-2a ((S,S)- or (R,R)-2a) forms a complementary duplex with an amidine dimer linked through the same linker amide chirality and sequence to that of 1a or 2a via salt bridges, leading to perfect chirality- and sequence-selective duplex formation (iv).
We anticipated that either a right- or left-handed homodouble helix would be induced in the carboxylic acid dimers when chiral linkers52, such as (S,S)- or (R,R)-trans-1,2-cyclohexane-based (cHex) bis-amide derivatives, were introduced between the monomer units (1 and 2, Fig. 1b). Incidentally, we have found a complete sequence-selective chiral self-sorting in a mixture of racemic (rac)-1a and rac-2a mediated by unique interstrand multihydrogen-bond-driven homoduplex formations, the structures of which are significantly different from our expected self-associated homodouble helices, and are further different from each other depending on the linker amide sequences (NHCO-cHex (L1) or CONH-cHex (L2), Fig. 1c). The further addition of an enantiopure amide-linked amidine dimer (3 or 4) with a particular linker amide sequence to a mixture of rac-1a and rac-2a results in the formation of a single optically pure complementary duplex with a 100% diastereoselectivity and sequence specificity stabilized by the amidinium–carboxylate salt bridges (Fig. 1c). In addition, we show that the diastereoselectivities during the complementary duplex formations, however, greatly rely on the sequence and chirality of the amide linkers of the amidine dimers relative to those of the carboxylic acid dimers and are almost independent of the amidine chirality. We also demonstrate a vital role of multihydrogen bonds formed between the amide linkers that determines the observed unique chirality- and sequence-selective homo- and complementary self-sorting.
A series of chiral dimers of carboxylic acids (1, 2) and amidines (3, 4) linked through chiral or meso amide residues with different sequences was prepared according to the reported methods (see Supplementary Methods)47,50,53.
Self-sorting behaviour of carboxylic acid dimers
We first investigated the self-association behaviour of the carboxylic acid dimers (R,R)- or (S,S)-1a and -2a. The circular dichroism (CD) spectra of the enantiomeric (R,R)- and (S,S)-strands of 1a and 2a in CDCl3 showed intense split-type Cotton effects that are mirror images of each other (Fig. 2a). However, the CD intensities significantly decreased in dimethyl sulfoxide (DMSO; Supplementary Fig. 1) because of dissociation into single strands in DMSO because DMSO is a strong hydrogen-bond acceptor and strongly hampers the interstrand hydrogen bonds; therefore, the CD spectral patterns and intensities of (R,R)-1a as well as (R,R)-2a in DMSO were similar to those of the carboxy-protected (R,R)-1b-OMe and -2b-OMe in CDCl3, respectively. The formation of homochiral duplexes 1a2 and 2a2 in CDCl3 was also supported by the corresponding dimer peaks [1a2+Na]+ and [2a2+Na]+ observed in their matrix-assisted laser desorption/ionization time-of-flight mass spectra (Supplementary Fig. 2).

(a) CD spectra (upper) of (S,S)-1a and -2a (0.50 mM) and (R,R)-1a and -2a (0.50 mM) in CDCl3 at 25 °C. Absorption spectra (bottom) of (S,S)-1a and -2a (0.50 mM) in CDCl3 at 25 °C are also shown. (b) Partial 1H NMR spectra of (S,S)-1a and -1b-OMe (1.0 mM) and (R,R)-2a and -2b-OMe (1.0 mM) in CDCl3 at 25 °C. The amide NH resonances were assigned using 1H 2D NMR spectroscopy (Supplementary Figs 11–13). (c,d) X-ray crystal structures of (S,S)-1a′ (c) and (S,S)-2a′ (d). Hydrogen atoms and solvent molecules are omitted for clarity. Black dotted lines represent interstrand hydrogen bonds. Average twist angles between the diphenylethynylene groups (red dotted lines) connecting to the amide linker, in which the twist angles are defined as the dihedral angles formed by the four carbon atoms (marked with red open circles), and interstrand hydrogen-bonding schemes are also shown.
The dimerization constants (Kd) of (R,R)-1a and (S,S)-2a in CHCl3 at 25 °C were then estimated to be ca. 3.2 × 107 and ca. 5.0 × 105M−1 by extrapolation of the Kd values measured in CHCl3/THF (tetrahydrofuran) and CHCl3/CH3CN mixtures with various ratios, respectively, because the Kd values in pure CHCl3 are too high to accurately estimate (Supplementary Fig. 3). These significantly higher Kd values as compared with those of the model compounds (R,R)-M1 and -M2 lacking the m-terphenyl moieties (Kd=ca. 20 M−1, Supplementary Fig. 4) and CC (Kd=ca. 1.4 × 103M−1)51 suggest the strong multiple interstrand hydrogen bonds, which were consistent with the significant downfield shifts of the amide NH proton resonances of (S,S)-1a and (R,R)-2a in their 1H NMR spectra in comparison with the corresponding carboxy-protected (S,S)-1b-OMe and (R,R)-2b-OMe (Fig. 2b). The variable-temperature 1H NMR spectral changes of (R,R)-1a and -2a also support the strong interstrand hydrogen bonds; the amide NH signals of both (R,R)-1a and -2a showed larger negative temperature coefficients (Δδ/ΔT (ppb per °C)) in DMSO-d6 (−5.8 and −4.9) than those in CDCl3 (−2.0 and −3.1; Supplementary Figs 5–8)54.
The X-ray crystallographic analyses of analogous (S,S)-1a′ and -2a′ whose structures are assumed to be almost identical to those of (S,S)-1a and -2a, respectively, except for the pendant and/or terminal substituents (see Fig. 1b), revealed a unique deeply intertwined duplex structure (Fig. 2c,d). Unexpectedly, these structures are completely different from a self-associated homodouble helix-like CC2 (see Fig. 1a). Each strand adopts a similar ‘U-shape’ structure resulting from the (S,S)-trans-1,2-cyclohexyl linker residue, binding together through the eight interstrand hydrogen bonds, in which each amide group at the linker moiety is sandwiched between the two carboxy groups of the other strand. The remarkable structural difference between (S,S)-1a′ and -2a′ in the solid state is that the homoduplex ((S,S)-2a′)2 has a well-packed structure due to the almost parallel orientation of the strands (Fig. 2d), while the intertwined strands of (S,S)-1a′ are oriented perpendicular to each other, resulting in a less sterically hindered structure (Fig. 2c). This difference in the steric hindrance between ((S,S)-1a′)2 and ((S,S)-2a′)2 is reasonably correlated with the difference in their self-association affinities; the Kd value of (S,S)-1a is ∼102 times greater than that of (S,S)-2a. The 1H two-dimensional (2D) nuclear Overhauser effect spectroscopy (NOESY) analysis of (R,R)-1a and (S,S)-2a in CDCl3 (Supplementary Figs 9–13) showed characteristic interstrand NOE cross-peaks including those between the linker cHex protons and the terminal aromatic protons of the m-terphenyl moieties along with the interstrand aromatic protons, indicating that such unique intertwined structures in the solid state were retained in solution. The observed large downfield shifts of the amide NH and CO2H protons of (S,S)-1a and (R,R)-2a in their 1H NMR spectra (Fig. 2b) also support the structures.
We next investigated the chiral self-sorting behaviour of rac-1a and rac-2a using 1H NMR in CDCl3 at 25 °C. The 1H NMR spectrum of rac-1a showed two sets of signals with an integral ratio of 2:1 (0% enantiomeric excess (e.e.), Fig. 3a), which did not coalesce even at 50 °C (Supplementary Fig. 14), whereas a single set of signals was observed in DMSO (Supplementary Fig. 15), in which the duplexes undergo the anticipated dissociation into the single strands. The NOESY spectrum of rac-1a in CDCl3 also exhibited interstrand NOEs similar to those for the enantiopure (R,R)-1a without chemical exchange cross-peaks between the two species except for the labile CO2H signals (Supplementary Figs 16 and 17), suggesting that the exchange rate between the species is slower than the present NMR timescale. The major set of signals of rac-1a is identical to the signals of the homochiral duplexes ((R,R)-1a)2 and ((S,S)-1a)2, and the minor set of the signals decreased with an increase in the % e.e. of 1a (Fig. 3a). Therefore, the minor signals are unambiguously assigned to the heterochiral duplex (R,R)-1a·(S,S)-1a.

(a) 1H NMR spectra of carboxylic acid CO2H and amide NH proton resonances of 1a (0.50 mM) with different % e.e. in CDCl3 at 25 °C. Blue and red circles denote the signals because of the homochiral and heterochiral duplexes, respectively. Full-scale spectra are shown in Supplementary Fig. 18a. (b) Plots of % d.e. of 1a2 versus the % e.e. of 1a in CDCl3 at 25 °C. The plots were fitted using Y=100{−2[4−3(X/100)2]0.5+5}/3 (equation A), where X and Y are % e.e. of 1a and % d.e. of 1a2, respectively (for details, see Supplementary Methods). (c) 1H NMR spectra of carboxylic acid CO2H and amide NH proton resonances of 2a (0.50 mM) with different % e.e. in CDCl3 at 25 °C. Full-scale spectra are shown in Supplementary Fig. 18b. (d) Partial 1H NMR spectra (0.50 mM) of (S,S)-2a, (S,S)-2c, an equimolar mixture (1.0 mM) of (S,S)-2a and (S,S)-2c, and an equimolar mixture (1.0 mM) of (R,R)-2a and (S,S)-2c in CDCl3 at 25 °C. Red circles denote the signals because of the duplex (S,S)-2a·(S,S)-2c. Full-scale spectra are shown in Supplementary Fig. 20.
The diastereomeric excess (d.e.) values (%) of 1a2 (homochiral duplex versus heterochiral duplex) were then plotted versus the % e.e. of 1a, which matched well with the simulated curve using the equation (Y=100{−2[4−3(X/100)2]0.5+5}/3) (inset in Fig. 3b; equation A) obtained by a modification of the reported equation (Fig. 3b)55,56 (for details, see Supplementary Methods), suggesting no preference in the diastereomeric duplex formation, thus forming the homochiral and heterochiral 1a2 duplexes in a 2:1 molar ratio in CDCl3 at 25 °C. As anticipated, however, the diastereomeric duplex formation is sensitive to temperature and its molar ratio changed on heating or cooling (Supplementary Fig. 14d), being attributed to a subtle change in the relative interstrand hydrogen-bonding strength between the homo- and heterochiral 1a2, which was in accordance with the observed differences in their amide NH temperature coefficients (Supplementary Fig. 14c).
In sharp contrast, rac-2a was completely chiral self-sorted to form only the homochiral duplexes ((R,R)-2a)2 and ((S,S)-2a)2, giving one set of 1H NMR signals in CDCl3 independent of the % e.e. of 2a (Fig. 3c) and temperature from −20 to 50 °C (Supplementary Fig. 19). This was also supported by the following mixing experiments of (S,S)- or (R,R)-2a with its trimethylsilyl (TMS)-deprotected analogue (S,S)-2c; an equimolar mixture of (S,S)-2a and (S,S)-2c in CDCl3 gave a new set of signals corresponding to the heteroduplex (S,S)-(2a)·(S,S)-2c, whereas no trace amount of such a distinguishable heteroduplex derived from (R,R)-2a and (S,S)-2c was observed for an equimolar mixture of (R,R)-2a and (S,S)-2c (Fig. 3d), indicating that rac-2a completely self-sorts to form only the homochiral duplexes ((R,R)-2a)2 and ((S,S)-2a). In addition, these observations exclude the possibility of the fast exchange between the hetero- and homochiral duplexes of rac-2a on the present NMR timescale.
The difference in the chiral self-sorting behaviour between 1a and 2a may be due to the difference in the twist angles between the two rigid diphenylethynylene groups connecting to the chiral amide linkers with a different sequence, in which the average twist angle in ((S,S)-2a′)2 (ca. 48°) is much larger than that in ((S,S)-1a′)2 (ca. 18°) in the solid state (Fig. 2c,d). The density functional theory (DFT) calculations revealed that the homochiral duplex ((S,S)-1a)2 is only 17.0 kJ mol−1 more stable than the heterochiral duplex (R,R)-1a·(S,S)-1a, whereas, for 2a, the homochiral duplex formation ((S,S)-2a)2 is much more favourable than the heterochiral duplex formation (R,R)-2a·(S,S)-2a by 51.7 kJ mol−1 (Supplementary Fig. 21a–d), being reasonably consistent with the previously discussed experimental results. The observed large energy difference (ΔE) between the homochiral and heterochiral duplexes for 2a2 (−51.7 kJ mol−1) as compared with that for 1a2 (−17.0 kJ mol−1) is attributed to a distorted structure of the heterochiral homoduplex (R,R)-2a·(S,S)-2a that lacks one interstrand hydrogen bond between the amide NH and carboxy C=O groups (Supplementary Fig. 21d), while all of the possible interstrand hydrogen bonds are retained in both the homo- and heterochiral 1a2 during the calculations because of the perpendicular orientation of the strands as seen in the solid state (Fig. 2c), resulting in a similar duplex structure (Supplementary Fig. 21a,b). Therefore, predominant homochiral self-sorting could not take place for rac-1a (Fig. 3a).
Of further interest is that an equimolar mixture of rac-1a and rac-2a showed complete self-sorting in terms of their linker amide sequences, giving only the enantiomeric pairs of the 1a2 and 2a2 duplexes as explicitly observed in their 1H NMR spectra in CDCl3 (Fig. 4a). Each duplex of 1a and 2a is unable to exchange, that is further supported by the 1H NMR and CD measurements of an equimolar mixture of (S,S)-1a and (S,S)-2a in CDCl3 (Fig. 4a,b); the 1H NMR spectrum of an equimolar mixture of (S,S)-1a and (S,S)-2a in CDCl3 displayed only two sets of the signals corresponding to ((S,S)-1a)2 and ((S,S)-2a)2 (Fig. 4a). Moreover, the mixture showed a CD spectrum precisely identical to the simulated one (Fig. 4b), indicating that the complete sequence-selective chiral self-sorting took place within mixtures of rac-1a and rac-2a. The DFT calculations demonstrate the important role of the interstrand hydrogen bonds; the energy-minimized duplex structure of (S,S)-1a·(S,S)-2a with the different linker amide sequences has a mismatched arrangement of the interstrand hydrogen bonds (Supplementary Fig. 21e), and is 11.1 and 32.6 kJ mol−1 less stable than those of the chiral self-sorted homoduplexes ((S,S)-2a)2 and ((S,S)-1a)2, respectively (Supplementary Fig. 21a,c). On the basis of the total energies of the calculated structures (Supplementary Fig. 21), the stabilities of the duplexes decrease in the following order: ((S,S)-1a)2>(R,R)-1a·(S,S)-1a>((S,S)-2a)2>(S,S)-1a·(S,S)-2a>(R,R)-2a·(S,S)-2a, which are in good agreement with the experimental results, and the energetically disfavoured duplexes (S,S)-1a·(S,S)-2a and (R,R)-2a·(S,S)-2a were not detected at all under the present experimental conditions.

(a) 1H NMR spectra of carboxylic acid CO2H and amide NH proton resonances of rac-1a (1.0 mM), rac-2a (1.0 mM), an equimolar mixture (1.0 mM) of rac-1a and rac-2a (S,S)-1a (0.50 mM), (S,S)-2a (0.50 mM) and an equimolar mixture (1.0 mM) of (S,S)-1a and (S,S)-2a in CDCl3 at 25 °C. Full-scale spectra are shown in Supplementary Fig. 22. (b) CD and absorption spectra of (S,S)-1a (0.50 mM), (S,S)-2a (0.50 mM) and those observed and simulated for the mixture of an equimolar amount of (S,S)-1a and (S,S)-2a (0.50 mM) in CDCl3 at 25 °C.
Chirality- and sequence-selective heteroduplex formation
With all the above results taken together with our previous findings of the complementary double-helix formations through amidinium–carboxylate salt bridges44,47,48,49,50, we envisaged that the optically active amidine dimers linked through the chiral or meso amide linkers with a different sequence (NHCO-cHex (L1) or CONH-cHex (L2)) (3a–e and 4a–c, Fig. 1b) could selectively recognize the carboxylic acid dimers (1a and 2a) according to the linker chirality and sequences, thereby leading to a unprecedented diastereo- and sequence-selective complementary duplex formation.
On mixing rac-2a and (R,R,R,R,R,R)-3a composed of the dimeric (R,R,R,R) amidine linked through the (R,R)-L2 linker in a 2:1 molar ratio in CDCl3, the 1H NMR spectrum immediately changed to that consisting of complementary diastereomeric duplexes of 3a·(R,R)-2a and 3a·(S,S)-2a together with the remaining 2a (Supplementary Fig. 25). The nonequivalent N–H proton signals appeared at a low magnetic field (13–13.5 p.p.m.), suggesting the preferred-handed duplex formation stabilized by salt bridges44,47,48,49,50. Because the chain exchange rate between the complementary dimer strands is slower than the NMR timescale49, the diastereoselectivity between (R,R,R,R,R,R)-3a and rac-2a was estimated to be d.e.=58% ((R,R)-2a-rich, run 1, Table 1) from their integral ratio, indicating the homochiral (R,R) selectivity with respect to their cHex linker chirality. This diastereoselectivity was further confirmed by CD; the observed CD spectrum is almost identical to the simulated CD (Supplementary Fig. 26).
Interestingly, the amidine dimer (R,R,S,S,R,R)-3b bearing the opposite (S,S)-L2 linker showed a perfect diastereoselectivity towards rac-2a, producing the complementary duplex of 3b·(S,S)-2a with d.e.>99% as well as (R,R)-2a (e.e.>99%) remaining as a homochiral duplex, as evidenced by the 1H NMR and identical experimental and simulated CD spectra (run 2, Table 1, Fig. 5a–c and Supplementary Figs 27 and 28). It should be noted that highly disfavoured 3b·(R,R)-2a was hardly formed even in the presence of a large excess of (R,R)-2a (12 equivalents), indicating that the binding affinity of (S,S)-2a to 3b is at least 105 times higher than that of (R,R)-2a (Supplementary Fig. 29). This surprisingly high diastereoselectivity57,58 of 3b towards rac-2a enabled the separation of rac-2a into the (S,S)- and (R,R)-2a enantiomers by facile silica gel column chromatography, giving the corresponding optically pure enantiomers in 85% and 63% yield, respectively (Supplementary Fig. 30). (R,R,S,S,R,R)-3b could be recovered for further separation of rac-2a. Moreover, the resolved optically pure (R,R)-2a (e.e.>99%) could be also used to separate rac-3b in principle. More practically, the chemical bonding of (R,R,S,S,R,R)-3b to chromatographic supports including silica gel will enable more efficient and preparative separation of rac-2a as well as other enantiomers as a novel chiral stationary phase (CSP) for chromatographic enantioseparation. The complementary 1:1 duplex formation of 3b·(S,S)-2a was further evidenced using an electron-spray ionization mass spectroscopic (ESI-MS) measurement and vapour pressure osmometry experiment (Supplementary Figs 31–33).

(a) Partial 1H NMR spectra of 3b (0.50 mM), rac-2a (0.50 mM), 3b·(R,R)-2a (0.50 mM), 3b·(S,S)-2a (0.50 mM) and a mixture of 3b (0.50 mM) and two equivalents of rac-2a (1.0 mM) in CDCl3 at 25 °C. Full-scale spectra are shown in Supplementary Fig. 27. (b) CD (upper) and absorption (bottom) spectra (0.50 mM) of 3b, (R,R)-2a, (S,S)-2a, 3b·(R,R)-2a and 3b·(S,S)-2a in CDCl3 at ambient temperature. (c) Experimental and simulated CD (d.e.=100%) and absorption spectra for a mixture of 3a (0.50 mM) and two equivalents of rac-2a (1.0 mM) in CDCl3 at ambient temperature. For the simulated CD and absorption spectra, see Supplementary Methods. (d) Partial 1H NMR spectra (0.5 mM) of 3b, 3b·(R,R)-2a and 3b·(S,S)-2a in CDCl3 at 25 °C. The linker amide NH resonances were assigned using 1H 2D NMR spectroscopy (Supplementary Fig. 62).
More interestingly, (R,R)-3c and (R,R)-3d, in which the chiral amidine residues were replaced by achiral isopropyl (3c) and cyclohexyl (3d) substituents, respectively, also performed a perfect diastereoselectivity towards rac-2a to form duplexes only with (R,R)-2a (d.e.>99%) having the same linker chirality, which also resulted in the optically pure (S,S)-2a (e.e.>99%) as the remaining homochiral duplex (runs 3, 4, Table 1 and Supplementary Figs 34–37), suggesting that the diastereoselective duplex formation is governed by the linker chirality, and the chiral amidine residues may be no longer required. In other words, an anticipated complementary double-stranded helix formation stabilized by chiral amidinium–carboxylate salt bridges that would occur on both sides of the linker may not be prerequisite in order to achieve diastereoselective duplex formations. Therefore, the amidine dimer having the chiral amidine residues linked through achiral meso-linker (R,R,meso,R,R)-3e completely lost its diastereoselectivity (d.e.=0%; run 5, Table 1 and Supplementary Figs 38 and 39).
Various attempts to obtain crystals of the complementary duplex dimers suitable for an X-ray analysis produced only amorphous solids. Therefore, the energy-minimized structures of the duplexes of (R,R,S,S,R,R)-3b·(S,S)-2a and (R,R,S,S,R,R)-3b·(R,R)-2a were constructed using the semiempirical molecular orbital calculations followed by the DFT calculations based on an analogous crystal structure47. The initial model structures of (R,R,S,S,R,R)-3b·(S,S)-2a and its diastereomer (R,R,S,S,R,R)-3b·(R,R)-2a with a right-handed twisted conformation, in which the pendant 1-octynyl groups are replaced by hydrogen atoms, were constructed so as to satisfy the following experimental results: (1) as shown in their 1H NMR spectra (Fig. 5d and Supplementary Fig. 62), all of the amide NH resonances of (R,R,S,S,R,R)-3b·(S,S)-2a showed significant downfield shifts compared with those of the amidine strand (R,R,S,S,R,R)-3b (Δδ=0.53 or 0.73 p.p.m.; Table 2, entry 2), indicating that all of the amide protons participate in hydrogen bonds. In contrast, the amide NH resonances of (R,R,S,S,R,R)-3b·(R,R)-2a were slightly shifted downfield (Δδ=0.12 or 0.09 p.p.m.), suggesting weak hydrogen bonds. (2) An analogous complementary dimeric duplex composed of the identical (R,R)-amidine and carboxylic acid dimer strands linked by diacetylene residues bound together through salt bridges was determined to have a right-handed helical structure using the single-crystal X-ray analysis47.
The resultant energy-minimized structures of (R,R,S,S,R,R)-3b·(S,S)-2a and (R,R,S,S,R,R)-3b·(R,R)-2a with their total energies are depicted in Fig. 6a,b, respectively, which revealed that they take a largely bent-shaped, right-handed double-helix-like structure, in which (R,R,S,S,R,R)-3b·(S,S)-2a is 57.4 kJ mol−1 more stable than the other. All of the linker amide protons in 3b·(S,S)-2a form inter- or intramolecular hydrogen bonds with the amide C=O groups (average NH···O distance=2.14 Å; Fig. 6a), whereas only two of the amide protons of 3b·(R,R)-2a participate in such hydrogen bonds with a longer average NH···O distance (2.30 Å; Fig. 6b). The observed difference in the hydrogen-bonding networks between 3b·(S,S)-2a and 3b·(R,R)-2a was supported by their 1H NMR spectra (Fig. 5d and Supplementary Fig. 62). Therefore, the calculated structures reasonably explain the present unexpectedly high diastereoselective duplex formation between 3b and (S,S)-2a. The 2D NOESY spectra of 3b·(S,S)-2a in CDCl3 showed interstrand NOE cross-peaks including those between the terminal TMS protons and the phenyl moieties of the amidine residues along with the interstrand aromatic protons, while interstrand NOEs between the linker cHex protons were not identified because of the same amide linker sequence (NHCO-cHex) whose chemical shifts were quite similar to each other (Supplementary Figs 68–70). On the other hand, an interstrand NOE was clearly observed between the linker cHex protons of 4b·(R,R)-2a because of the different linker amide sequences (Supplementary Figs 71 and 72). Considering all the results including the 2D NOESY, ESI-MS, vapour pressure osmometry, salt-bridge formations and large downfield shifts of the linker amide NH resonances of 3b·(S,S)-2a, the calculated structure (Fig. 6a) is most likely retained in solution.

Capped-stick drawings of the structures for the homochiral duplex (R,R,S,S,R,R)-3b·(S,S)-2a (a) and the heterochiral duplex (R,R,S,S,R,R)-3b·(R,R)-2a (b) with respect to the cHex linker chirality optimized by DFT calculations. DFT-calculated energies are also shown in the bottom.
The amidine dimers 3a–3d also formed a duplex in a diastereoselective manner towards rac-1a with the linker amide sequence (NHCO-cHex) different from that of rac-2a and 3a–3d (CONH-cHex; runs 6–9, Table 1 and Supplementary Figs 40–47). However, their diastereoselectivities significantly decreased with the opposite heterochiral selectivity with respect to the cHex linker chirality, for example, (R,R)-3c preferentially formed a duplex with (S,S)-1a in 64% d.e., which is in significant contrast to the homochiral selectivity observed between 3a–3d and rac-2a (runs 1–4, Table 1). Again, the linker chirality plays a critical role so that (R,R,meso,R,R)-3e showed no diastereoselectivity (run 10, Table 1 and Supplementary Figs 48 and 49). The reason for this heterochiral preference between 3a-d and rac-1a is not totally understood, but may be due to the difference in the interstrand hydrogen-bonding networks that could be more efficiently formed between the heterochiral linker amide residues than between the homochiral counterparts. This speculation is supported by the fact that the amide NH resonances of, for instance, the heterochiral (R,R,S,S,R,R)-3b·(R,R)-1a showed higher downfield shifts (Δδ=0.56 p.p.m.) as compared with those of the homochiral (R,R,S,S,R,R)-3b·(S,S)-1a (Δδ=0.03 p.p.m.), respectively (Supplementary Fig. 64 and Table 2, entry 1).
We also investigated the diastereoselective duplex formations using a series of amidine dimers (4a–4c) with the NHCO-cHex linker amide sequence towards rac-1a and rac-2a. In all the duplex formations, moderate diastereoselectivities (64–80% d.e.) were observed except for that between 4b and rac-2a (34% d.e.; runs 11–16, Table 1 and Supplementary Figs 50–61), which were in contrast to the perfect diastereoselectivities achieved between 3b–3d and rac-2a. As for the selectivities with regard to the cHex amide linker chirality, there is the same tendency; 4a–4c favourably formed duplexes with 1a and 2a with the same and opposite configurations, respectively. As a typical example, (R,R)-4c preferentially formed a duplex with (R,R)-1a, but with (S,S)-2a in 70 and 80% d.e., respectively. The observed diastereoselectivities also rely on the difference in the interstrand hydrogen-bond strengths between the diastereomeric duplexes as revealed by more downfield shifts of the amide NH protons for the major duplex diastereoselectively formed during the complexations (Supplementary Figs 65–67 and Table 2, entries 3 and 4).
On the basis of these results, we anticipated that particular chiral amidine dimers would simultaneously recognize the chirality and sequence within a mixture of complementary carboxylic acid dimers via specific duplex formations. In fact, the mixing of two equivalents of each of rac-1a and rac-2a with (R,R,S,S,R,R)-3b in CDCl3 resulted in the formation of only the (R,R,S,S,R,R)-3b·(S,S)-2a duplex (d.e.>99%), while no duplex formation was observed towards rac-1a. As a result, an optically pure (R,R)-2a (e.e.>99%) quantitatively remained as a homochiral duplex together with free rac-1a (Fig. 7); thus, the perfect sequence- (CONH-cHex or NHCO-cHex) and chirality- ((R,R) or (S,S)) selective duplex formation was achieved as unambiguously evidenced using their 1H NMR analysis (Fig. 7a) and the observed and simulated CD spectra (Fig. 7b,c). The linker chirality rather than the amidine chirality also plays a vital role in the sequence- and chirality-selective complementary duplex formation; therefore, (R,R)-3d composed of achiral amidine residues formed a duplex only with (R,R)-2a with a complete diastereoselectivity (d.e.>99%) and sequence specificity in a mixture of rac-1a and rac-2a (Supplementary Figs 74 and 75).

(a) Partial 1H NMR spectra of rac-2a (0.50 mM), rac-1a (0.50 mM), 3b·(R,R)-2a (0.50 mM), 3b·(S,S)-2a (0.50 mM), 3b·(R,R)-1a (0.50 mM), 3b·(S,S)-1a (0.50 mM) and a mixture of 3b (0.50 mM) and two equivalents of rac-2a (1.0 mM) and rac-1a (1.0 mM) in CDCl3 at 25 °C. Full-scale spectra are shown in Supplementary Fig. 73. (b) CD (upper) and absorption (bottom) spectra (0.50 mM) of 3b·(S,S)-2a, (R,R)-2a and rac-1a in CDCl3 under ambient temperature. (c) Experimental and simulated CD (d.e.=100%) and absorption spectra for a mixture of 3b (0.50 mM) and two equivalents of each of rac-1a (1.0 mM) and rac-2a (1.0 mM) in CDCl3 under ambient temperature.
Discussion
We demonstrate here an unprecedented successive chiral self-sorting during the unique homo- and subsequent heteroduplex formations through interstrand multihydrogen bonds that take place in a perfect sequence-selective way accompanied with an extraordinary high diastereoselectivity in the latter case, which enables the separation of the racemic strands into the enantiomers. The present chirality- and sequence-selective successive self-sorting is most likely achieved because of the rigid chiral geometries generated by the chiral amide-linked dimeric strands that arrange functional groups in a suitable way for specific duplex formations.
In our previous studies, we showed that a carboxylic acid dimer such as CC formed an intertwined homodouble helix via interstrand association of the carboxylic acids51, which further formed a double-stranded helix (AA·CC) with the complementary amidine dimer such as AA (Fig. 1a)44,47,48,49. The helical handedness of the complementary double helix is fully controlled by the chirality introduced on the amidine residues49. On the basis of the present studies, however, it appears that the chirality and sequence (NHCO-cHex or CONH-cHex) of the amide linkers of dimeric carboxylic acid and amidine strands are of primary importance and dictate the overall chirality- and sequence-selective self-assemblies of the dimer strands, resulting from the unique multihydrogen-bonding networks formed between the linker amide residues. Importantly, the amidine chirality is not almost involved in the present diastereoselective complementary duplex formations that mostly rely on the sequence and chirality of the amide linkers of the amidine dimers relative to those of the carboxylic acid dimers.
The dimers of amidines and carboxylic acids possess reactive trimethylsilylethynyl groups at the ends, which allows longer oligomers joined by chiral amide linkers with specific sequence and chirality to be synthesized, which would provide a unique strategy for asymmetric template synthesis and an artificial replication system59,60 based on chirality- and sequence-selective multihydrogen-bond-assisted duplex formations, and also provide a clue towards a better understanding of the biological chiral self-sorting process.
Methods
General procedures for the complementary duplex formations
A typical procedure for the diastereoselective duplex formations between amidine and carboxylic acid dimers is described below. Stock solutions of (R,R,R,R,R,R)-3a (2.0 mM; solution I) and rac-2a (2.0 mM; solution II) were prepared in dry CDCl3. Aliquots of I (0.40 μmol, 200 μl), II (0.80 μmol, 400 μl) and dry CDCl3 (200 μl) were added to an NMR tube, and its 1H NMR spectrum was measured at 25 °C to determine the % d.e. of the 3a·2a duplexes (Supplementary Fig. 25). The solution was also used for measuring the CD (Supplementary Fig. 26). In a similar way, other diastereoselective duplex formations were preformed and their % d.e. values were estimated (Table 1).
Optical resolution of rac-2a via the heteoduplex formation
(R,R,S,S,R,R)-3b (5.44 mg, 3.58 μmol) and two equivalents of rac-2a (9.46 mg, 7.16 μmol) were dissolved in CHCl3 (2 ml), which produced an equimolar mixture of 3b·(S,S)-2a and (R,R)-2a judging from its 1H NMR spectrum (Fig. 5a) and were separated into the first and second fractions, respectively, using flash column chromatography (SiO2, 2 cm (i.d.) × 12 cm; eluent; CHCl3/MeOH=1/0–50/1, v/v). The % d.e. of the 3b·(S,S)-2a duplex obtained (6.3 mg) was >99% as estimated using its CD and 1H NMR spectra (Supplementary Fig. 30a,c)), while the second fraction mainly contained (R,R)-2a along with a small amount of 3b·(S,S)-2a and an unknown compound probably generated during the column chromatography. Thus, the second fraction was purified again using flush column chromatography, yielding 3b·(S,S)-2a (2.3 mg, >99% d.e., total yield; 8.6 mg, 85%) and (R,R)-2a containing a small amount of the unknown compound. Further purification using recycle size-exclusion chromatography (eluent; CHCl3) afforded pure (R,R)-2a (2.99 mg) in 63% yield; its e.e. value was estimated to be >99% on the basis of the CD and 1H NMR measurements (Supplementary Fig. 30b,c).
Additional information
Accession codes: The X-ray crystallographic coordinates for structures ((S,S)-1a′ and (S,S)-2a′) reported in this Article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 1036590 and 1036591. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
How to cite this article: Makiguchi, W. et al. Chirality- and sequence-selective successive self-sorting via specific homo- and complementary-duplex formations. Nat. Commun. 6:7236 doi: 10.1038/ncomms8236 (2015).
References
Berg, J. M., Tymoczko, J. L. & Stryer, L. Biochemistry 6th ed W. H. Freeman (2007).
Kuriyan, J., Konforti, B. & Wemmer, D. The Molecules of Life: Physical and Chemical Principles Garland Science - Taylor & Francis Group (2012).
Rowan, A. E. & Nolte, R. J. M. Helical molecular programming. Angew. Chem. Int. Ed. 37, 63–68 (1998).
Milton, R. C. D., Milton, S. C. F. & Kent, S. B. H. Total chemical synthesis of a D-enzyme: the enantiomers of HIV-1 protease show demonstration of reciprocal chiral substrate-specificity. Science 256, 1445–1448 (1992).
Mandal, K. et al. Chemical synthesis and X-ray structure of a heterochiral {D-protein antagonist plus vascular endothelial growth factor} protein complex by racemic crystallography. Proc. Natl Acad. Sci. USA 109, 14779–14784 (2012).
Garbesi, A. et al. L-DNAs as potential antimessenger oligonucleotides: a reassessment. Nucleic Acids Res. 21, 4159–4165 (1993).
Krämer, R., Lehn, J.-M. & Marquis-Rigault, A. Self-recognition in helicate self-assembly: spontaneous formation of helical metal complexes from mixtures of ligands and metal ions. Proc. Natl Acad. Sci. USA 90, 5394–5398 (1993).
Caulder, D. L. & Raymond, K. N. Supramolecular Self-recognition and self-assembly in gallium(III) catecholamide triple helices. Angew. Chem. Int. Ed. Engl. 36, 1440–1442 (1997).
Albrecht, M., Schneider, M. & Röttele, H. Template-directed self-recognition of alkyl-bridged bis(catechol) ligands in the formation of helicate-type complexes. Angew. Chem. Int. Ed. 38, 557–559 (1999).
Wu, A. & Isaacs, L. Self-sorting: the exception or the rule? J. Am. Chem. Soc. 125, 4831–4835 (2003).
Addicott, C., Das, N. & Stang, P. J. Self-recognition in the coordination driven self-assembly of 2-D polygons. Inorg. Chem. 43, 5335–5338 (2004).
Cao, L.-P., Wang, J.-G., Ding, J.-Y., Wu, A.-X. & Isaacs, L. Reassembly self-sorting triggered by heterodimerization. Chem. Commun. 47, 8548–8550 (2011).
Jiménez, A. et al. Selective encapsulation and sequential release of guests within a self-sorting mixture of three tetrahedral cages. Angew. Chem. Int. Ed. 53, 4556–4560 (2014).
Talotta, C., Gaeta, C., Qi, Z., Schalley, C. A. & Neri, P. Pseudorotaxanes with self-sorted sequence and stereochemical orientation. Angew. Chem. Int. Ed. 52, 7437–7441 (2013).
Ayme, J.-F., Beves, J. E., Campbell, C. J. & Leigh, D. A. The self-sorting behavior of circular helicates and molecular knots and links. Angew. Chem. Int. Ed. 53, 7823–7827 (2014).
Masood, M. A., Enemark, E. J. & Stack, T. D. P. Ligand self-recognition in the self-assembly of a [{Cu(L)}2]2+ complex: the role of chirality. Angew. Chem. Int. Ed. 37, 928–932 (1998).
Prins, L. J., Huskens, J., de Jong, F., Timmerman, P. & Reinhoudt, D. N. Complete asymmetric induction of supramolecular chirality in a hydrogen-bonded assembly. Nature 398, 498–502 (1999).
Telfer, S. G., Bernardinelli, G. & Williams, A. F. Diastereospecific synthesis of amino-acid substituted 2,2’-bipyridyl complexes. Chem. Commun. 1498–1499 (2001).
Shi, X., Fettinger, J. C. & Davis, J. T. Homochiral G-quadruplexes with Ba2+ but not with K+: the cation programs enantiomeric self-recognition. J. Am. Chem. Soc. 123, 6738–6739 (2001).
Ishida, Y. & Aida, T. Homochiral supramolecular polymerization of an “S”-shaped chiral monomer: translation of optical purity into molecular weight distribution. J. Am. Chem. Soc. 124, 14017–14019 (2002).
Chung, D. M. & Nowick, J. S. Enantioselective molecular recognition between β-sheets. J. Am. Chem. Soc. 126, 3062–3063 (2004).
Hwang, I.-W. et al. Porphyrin boxes constructed by homochiral self-sorting assembly: optical separation, exciton coupling, and efficient excitation energy migration. J. Am. Chem. Soc. 126, 16187–16198 (2004).
Telfer, S. G., Sato, T. & Kuroda, R. Noncovalent ligand strands for transition-metal helicates: the straightforward and stereoselective self-assembly of dinuclear double-stranded helicates using hydrogen bonding. Angew. Chem. Int. Ed. 43, 581–584 (2004).
Hutin, M. et al. Self-sorting chiral subcomponent rearrangement during crystallization. J. Am. Chem. Soc. 129, 8774–8780 (2007).
Lisowski, J. Enantiomeric self-recognition in homo- and heterodinuclear macrocyclic lanthanide(III) complexes. Inorg. Chem. 50, 5567–5576 (2011).
Safont-Sempere, M. M. et al. Impact of molecular flexibility on binding strength and self-sorting of chiral π-surfaces. J. Am. Chem. Soc. 133, 9580–9591 (2011).
Helmich, F., Smulders, M. M. J., Lee, C. C., Schenning, A. P. H. J. & Meijer, E. W. Effect of stereogenic centers on the self-sorting, depolymerization, and atropisomerization kinetics of porphyrin-based aggregates. J. Am. Chem. Soc. 133, 12238–12246 (2011).
Ponnuswamy, N., Cougnon, F. B. L., Clough, J. M., Pantoş, G. D. & Sanders, J. K. M. Discovery of an organic trefoil knot. Science 338, 783–785 (2012).
Lin, N.-T. et al. Enantioselective self-sorting on planar, π-acidic surfaces of chiral anion-π transporters. Chem. Sci. 3, 1121–1127 (2012).
Roche, C. et al. Homochiral columns constructed by chiral self-sorting during supramolecular helical organization of hat-shaped molecules. J. Am. Chem. Soc. 136, 7169–7185 (2014).
Gütz, C. et al. Enantiomerically pure trinuclear helicates via diastereoselective self-assembly and characterization of their redox chemistry. J. Am. Chem. Soc. 136, 11830–11838 (2014).
Safont-Sempere, M. M., Fernández, G. & Würthner, F. Self-sorting phenomena in complex supramolecular systems. Chem. Rev. 111, 5784–5814 (2011).
Saha, M. L. & Schmittel, M. Degree of molecular self-sorting in multicomponent systems. Org. Biomol. Chem. 10, 4651–4684 (2012).
Raynal, M., Ballester, P., Vidal-Ferran, A. & van Leeuwen, P. W. N. M. Supramolecular catalysis. Part 2: artificial enzyme mimics. Chem. Soc. Rev. 43, 1734–1787 (2014).
Hoeben, F. J. M., Jonkheijm, P., Meijer, E. W. & Schenning, A. P. H. J. About supramolecular assemblies of π-conjugated systems. Chem. Rev. 105, 1491–1546 (2005).
Hembury, G. A., Borovkov, V. V. & Inoue, Y. Chirality-sensing supramolecular systems. Chem. Rev. 108, 1–73 (2008).
Zhan, C., Léger, J.-M. & Huc, I. Cross-hybridization of pyridinedicarboxamide helical strands and their N-oxides. Angew. Chem. Int. Ed. 45, 4625–4628 (2006).
Kitamura, M., Yamakawa, M., Oka, H., Suga, S. & Noyori, R. Homochiral and heterochiral dimers of the methylzinc alkoxide formed from dimethylzinc and enantiomeric 3-exo-(dimethylamino)isoborneol—origin of the distinct differences in solution-phase behavior and crystal structures. Chem. Eur. J. 2, 1173–1181 (1996).
Wu, A., Chakraborty, A., Fettinger, J. C., Flowers, R. A. II & Isaacs, L. Molecular clips that undergo heterochiral aggregation and self-sorting. Angew. Chem. Int. Ed. 41, 4028–4031 (2002).
Claessens, C. G. & Torres, T. Chiral self-discrimination in a M3L2 subphthalocyanine cage. J. Am. Chem. Soc. 124, 14522–14523 (2002).
Satyanarayana, T., Abraham, S. & Kagan, H. B. Nonlinear effects in asymmetric catalysis. Angew. Chem. Int. Ed. 48, 456–494 (2009).
Jacquws, J., Collet, A. & Wilen, S. H. Enantiomers, racemates, and resolutions Krieger Publishing Company (1994).
Nowick, J. S. & Chung, D. M. Sequence-selective molecular recognition between β sheets. Angew. Chem. Int. Ed. 42, 1765–1768 (2003).
Ito, H., Furusho, Y., Hasegawa, T. & Yashima, E. Sequence- and chain-length-specific complementary double-helix formation. J. Am. Chem. Soc. 130, 14008–14015 (2008).
Wittung, P., Nielsen, P. E., Buchardt, O., Egholm, M. & Nordén, B. DNA-like double helix formed by peptide nucleic acid. Nature 368, 561–563 (1994).
Marquis, A. et al. Messages in molecules: Ligand/cation coding and self-recognition in a constitutionally dynamic system of heterometallic double helicates. Chem. Eur. J. 12, 5632–5641 (2006).
Tanaka, Y., Katagiri, H., Furusho, Y. & Yashima, E. A modular strategy to artificial double helices. Angew. Chem. Int. Ed. 44, 3867–3870 (2005).
Yashima, E., Maeda, K. & Furusho, Y. Single- and double-stranded helical polymers: synthesis, structures, and functions. Acc. Chem. Res. 41, 1166–1180 (2008).
Yamada, H., Wu, Z.-Q., Furusho, Y. & Yashima, E. Thermodynamic and kinetic stabilities of complementary double helices utilizing amidinium-carboxylate salt bridges. J. Am. Chem. Soc. 134, 9506–9520 (2012).
Maeda, T. et al. Double-stranded helical polymers consisting of complementary homopolymers. J. Am. Chem. Soc. 130, 7938–7945 (2008).
Makiguchi, W., Kobayashi, S., Furusho, Y. & Yashima, E. Formation of a homo double helix of a conjugated polymer with carboxy groups and amplification of the macromolecular helicity by chiral amines sandwiched between the strands. Angew. Chem. Int. Ed. 52, 5275–5279 (2013).
Makiguchi, W. et al. Homo-double helix formation of an optically active conjugated polymer bearing carboxy groups and amplification of the helicity upon complexation with achiral and chiral amines. J. Polym. Sci. 53, 990–999 (2015).
Yamada, H., Furusho, Y. & Yashima, E. Diastereoselective imine-bond formation through complementary double-helix formation. J. Am. Chem. Soc. 134, 7250–7253 (2012).
Cierpicki, T. & Otlewski, J. Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 21, 249–261 (2001).
Luchinat, C. & Roelens, S. Enantiomeric purity determination of 1,2-diols through NMR spectroscopy without chiral auxiliaries. J. Am. Chem. Soc. 108, 4873–4878 (1986).
Safont-Sempere, M. M., Osswald, P., Radacki, K. & Würthner, F. Chiral self-recognition and self-discrimination of strapped perylene bisimides by π-stacking dimerization. Chem. Eur. J. 16, 7380–7384 (2010).
Kubo, Y., Maeda, S., Tokita, S. & Kubo, M. Colorimetric chiral recognition by a molecular sensor. Nature 382, 522–524 (1996).
Levkin, P. A., Maier, N. M., Schurig, V. & Lindner, W. Strong detrimental effect of a minute enantiomeric impurity of a chiral selector on the enantioselectivity factor. Angew. Chem. Int. Ed. 49, 7742–7744 (2010).
Lindsey, J. S. Self-assembly in synthetic routes to molecular devices. Biological principles and chemical perspectives: a review. New J. Chem. 15, 153–180 (1991).
Robertson, A., Sinclair, A. J. & Philp, D. Minimal self-replicating systems. Chem. Soc. Rev. 29, 141–152 (2000).
Acknowledgements
This work was supported in part by a Grant-in-Aids for Scientific Research (S) from the Japan Society for the Promotion of Science (JSPS; no. 25220804) and by the Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology, Japan. W.M. and J.T. express thanks for a JSPS Research Fellowship for Young Scientists (no. 480 for W.M. and no. 8886 for J.T.).
Author information
Authors and Affiliations
Contributions
E.Y. designed and directed the project, and W.M., J.T. and H.Y. performed the experiments. H.I. confirmed the validity of the X-ray crystallographic analysis. N.O. calculated the duplex structures by DFT. All authors analysed the data and discussed the results. N.O. and E.Y. co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-117, Supplementary Tables 1-2, Supplementary Methods and Supplementary References (PDF 12791 kb)
Supplementary Data 1
cif file for x-ray crystal structure of (S,S)-1a' (CIF 73 kb)
Supplementary Data 2
cif file for x-ray crystal structure of (S,S)-2a' (CIF 74 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Makiguchi, W., Tanabe, J., Yamada, H. et al. Chirality- and sequence-selective successive self-sorting via specific homo- and complementary-duplex formations. Nat Commun 6, 7236 (2015). https://doi.org/10.1038/ncomms8236
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms8236
This article is cited by
-
Temperature-controlled repeatable scrambling and induced-sorting of building blocks between cubic assemblies
Nature Communications (2019)
-
Chiral self-sorting process in the self-assembly of homochiral coordination cages from axially chiral ligands
Communications Chemistry (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
