
Abstract
Nature has created [FeFe]-hydrogenase enzyme as a hydrogen-forming catalyst with a high turnover rate. However, it does not meet the demands of economically usable catalytic agents because of its limited stability and the cost of its production and purification. Synthetic chemistry has allowed the preparation of remarkably close mimics of [FeFe]-hydrogenase but so far failed to reproduce its catalytic activity. Most models of the active site represent mimics of the inorganic cofactor only, and the enzyme-like reaction that proceeds within restricted environments is less well understood. Here we report that chitosan, a natural polysaccharide, improves the efficiency and durability of a typical mimic of the diiron subsite of [FeFe]-hydrogenase for photocatalytic hydrogen evolution. The turnover number of the self-assembling system increases ~4,000-fold compared with the same system in the absence of chitosan. Such significant improvements to the activity and stability of artificial [FeFe]-hydrogenase-like systems have, to our knowledge, not been reported to date.
Similar content being viewed by others


Introduction
Enzymes may bind substrates through multiple interactions in elaborate pockets to direct a specific reaction pathway under mild conditions1,2,3,4. [FeFe]-hydrogenase ([FeFe]-H2ase)5,6, a natural enzyme for hydrogen (H2) evolution, is deeply embedded within the protein matrix to enable the reversible reduction of protons to H2 with low overpotential and high turnover frequencies (TOF 6,000–9,000 s−1 per catalytic site). The high-resolution X-ray crystallographic structures establish that [FeFe]-H2ase, isolated from Desulfovibrio desulfuricans5 and Clostridium pasteurianum6, features a butterfly [Fe2S2] subunit coordinated by a cysteine-linked [Fe4S4] cluster, carbon monoxide and cyanide ligands, and by a dithiolate bridging the two iron centres. The diiron [Fe2S2] subunit serves as the catalytic centre for proton reduction, and the [Fe4S4] cluster mediates transfer electron to and from the active site of the H-cluster. The astonishing rates of H2 production from the non-precious diiron catalysts via a group of enzymes under mild conditions can exceed those of platinum. However, the large-scale isolation of the enzyme from natural systems is rather difficult, hence the development of artificial [FeFe]-H2ase analogues capable of reproducing the enzymic activity has spurred considerable interest in both the scientific and industrial communities7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25. Over the past decade, a variety of mimics of the diiron subsite of [FeFe]-H2ase have been shown to function as catalysts for chemical reduction of protons26,27,28,29,30,31,32,33. It has been clear that electron transfer, either electrochemical or photochemical, to a mimic of the active site of [FeFe]-H2ase is a prerequisite for H2 evolution10,11,12,13,14,22. From a photochemical point of view, the electron transfer is triggered by the absorption of a photon by a photosensitizer13,14,15,16,17,18,19,20,21,22,23,24,25. Since the first attempt by Sun and Åkermark34 to construct an artificial photocatalytic system for H2 evolution in 2003, a large number of synthetic model complexes have been pursued to mimic the structure and functionality of the diiron subunit of the natural [FeFe]-H2ase H-cluster35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51. It is encouraging to see that the catalytic efficiency for H2 evolution from artificial photocatalytic systems using mimics of the diiron subsite of [FeFe]-H2ase as catalysts has been increased from null to more than hundreds or thousands of turnover numbers (TON) under different irradiation conditions. In comparison to the efficient diiron active site of [FeFe]-H2ase in nature, however, no [FeFe]-H2ase mimic has been able to duplicate the high level of reactivity of natural [FeFe]-H2ase. Review of the literature indicates that the synthetic mimics of [FeFe]-H2ase reported thus far are mainly focused on the inorganic cofactor only, and the enzyme-like reaction that proceeds within restricted environments is to date poorly understood.
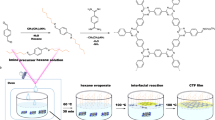
With this in mind, we initiated the study of a chitosan-confined mimic of the diiron subsite of [FeFe]-H2ase for H2 production. Chitosan is a naturally occurring polysaccharide containing a significant content of primary amines and hydroxyl groups52,53,54. When the amines are protonated by acids, chitosan bears a polycationic character. In view of the chelation and electrostatic interactions, we envision that chitosan may incorporate mimics of the diiron subsite of [FeFe]-H2ases intimately, as is the case of [FeFe]-H2ase, which is buried deeply within the protein matrix in nature. To avoid side-chain effects, the simplest mimic of the diiron subsite of [FeFe]-H2ases, [Fe2(CO)6(μ-adt)CH2C6H5] [μ-adt=N(CH2S)2]27,37, is selected as a catalyst (Fig. 1). The 3-mercaptopropionic acid (MPA)-capped CdTe quantum dots (MPA-CdTe QDs), promising for H2 evolution in combination with a mimic of the diiron subsite of [FeFe]-H2ase (ref. 45), are used as the photosensitizer. Herein, CdTe QDs are stabilized by MPA and their negatively charged surfaces55 preferably interact with cationic chitosan. Ascorbic acid (H2A) serves as not only a proton source to protonate the amines of chitosan and the catalytic intermediate of photoreduced mimic of the diiron subsite of [FeFe]-H2ase but also as a sacrificial electron donor to regenerate MPA-CdTe QDs for photocatalytic H2 production. Significantly, the self-assembled system that comprises chitosan, [Fe2(CO)6(μ-adt)CH2C6H5], MPA-CdTe QDs and H2A is capable of producing H2 with TON of up to (5.28±0.17) × 104 and initial TOF of 1.40±0.22 s−1 with respect to [Fe2(CO)6(μ-adt)CH2C6H5] catalyst under visible light irradiation (λ>400 nm). The catalytic stability is enhanced from 8 to 60 h and the catalytic activity is over 4.16 × 103-fold higher than that of the same system without chitosan. The activity and stability are, to the best of our knowledge, the highest to date for light-driven catalytic H2 evolution from mimics of the diiron subsite of [FeFe]-H2ase.

A schematic describing the H2 photogeneration of a chitosan-confined mimic of the diiron subsite of [FeFe]-H2ase in the presence of CdTe quantum dots and H2A.
Results
The photocatalytic activity of H2 evolution
An initial photocatalytic experiment of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst with MPA-CdTe QDs was evaluated in the absence of chitosan. To keep the solubility of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst throughout the experiment, we carried out the reaction in a mixture of methanol and water. The anaerobic solution, containing [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (1.00 × 10−5 mol l−1), MPA-CdTe QDs (0.86 × 10−6 mol l−1), along with H2A (0.10 mol l−1), was irradiated by light-emitting diodes (λ=410 nm) at room temperature, where the best ratio of methanol to water was found to be 1:3 (v-v) (Supplementary Fig. S1). The photoproduct of H2 was characterized by gas chromatography (GC) analysis with methane as the internal standard. The time course showed that the amount of H2 increased in the first 4 h and then leveled off, yielding a TON of only 1.74±0.06 based on [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (Fig. 2a, line A). In sharp contrast, the catalytic performance of the same solution was improved significantly in the presence of 1.0 g l−1 of chitosan. Line B in Fig. 2a shows the H2 production over time from the mixture under visible light irradiation. The amount of H2 reached 1.27±0.01 ml (TON=569±2) within 10 h of irradiation, and the rate of H2 evolution was almost linear even after 10 h of irradiation. Control experiments further proved that the components in the system, [Fe2(CO)6(μ-adt)CH2C6H5] catalyst, MPA-CdTe QDs, H2A, chitosan or light are all essential for efficient H2 generation. The absence of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst led to the rate of H2 evolution dropping dramatically and no H2 could be detected when either MPA-CdTe QDs or H2A was absent from the reaction system with chitosan (Supplementary Fig. S2). The difference in the catalytic activity (1.74 versus 569) and stability (4 versus 10 h) for the systems with and without chitosan under the same condition implies that chitosan has a key role in the photocatalytic H2 evolution.

(a) H2 evolution in the absence (A) and presence (B) of chitosan (1.0 g l−1), containing MPA-CdTe QDs (0.86 × 10−6 mol l−1), [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (1.00 × 10−5 mol l−1) and H2A (0.10 mol l−1) in methanol/water (1:3 v-v); (b) H2 evolution as a function of chitosan concentrations, containing MPA-CdTe QDs (0.86 × 10−6 mol l−1), [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (1.00 × 10−5 mol l−1) and H2A (0.10 mol l−1) in methanol/water (1:3 v-v); (c) H2 evolution at various pH values, containing MPA-CdTe QDs (0.86 × 10−6 mol l−1), [Fe2(CO)6(μ-adt)CH2C6H5] (1.00 × 10−5 mol l−1), H2A (0.10 mol l−1) and chitosan (1.0 g l−1) in methanol/water (1:3 v-v); (d) H2 evolution under the optimized conditions in the absence (A) and presence (B) of chitosan, containing MPA-CdTe QDs (1.71 × 10−6 mol l−1), [Fe2(CO)6(μ-adt)CH2C6H5] (1.00 × 10−6 mol l−1) and H2A (0.20 mol l−1) in methanol/water (1:3 v-v) at pH 4.5. Error bars represent mean±s.d. of parallel experiments.
Furthermore, the amounts of chitosan together with MPA-CdTe QDs and [Fe2(CO)6(μ-adt)CH2C6H5] catalyst were carefully investigated to optimize the reaction. Note that 1.0 g l−1 chitosan is the best concentration to achieve the highest TON of the assembled system for H2 evolution under a given pH condition (Fig. 2b). And the smaller size of MPA-CdTe QDs that is emissive at shorter wavelength gives rise to the higher TON for the photocatalytic H2 evolution system (Supplementary Fig. S3). The highest TON was obtained in the presence of MPA-CdTe QDs Green (2.8 nm) (Supplementary Table S1). As the conduction band energy of MPA-CdTe QDs Green is over −2.0 V, we could detect a small amount of H2 from the system without catalyst (Supplementary Fig. S2). Under the same condition, that is, 10 ml methanol/water solution (1:3, v-v) containing chitosan (1.0 g l−1), MPA-CdTe QDs (0.86 × 10−6 mol l−1) and H2A (0.10 mol l−1) at pH 4.0, the amount of H2 was 6.61±0.48 μl per 10 h in the absence of catalyst. However, the presence of catalyst, [Fe2(CO)6(μ-adt)CH2C6H5] (1.00 × 10−5 mol l−1), resulted in H2 evolution efficiently (1.27±0.01 ml per 10 h). Moreover, the rate of H2 evolution increased as a function of the concentration of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (Supplementary Fig. S4). When the concentration of the catalyst was higher than 1.00 × 10−5 mol l−1, where the ideal concentration of MPA-CdTe QDs was 1.71 × 10−6 mol l−1 (Supplementary Fig. S5), the rate of H2 evolution would be no longer linear. In this situation, the highest TON value was achieved at 1.00 × 10−6mol l−1 of [Fe2(CO)6(μ-adt)CH2C6H5] (Supplementary Fig. S4), the optimal ratio of MPA-CdTe QDs to [Fe2(CO)6(μ-adt)CH2C6H5] catalyst is therefore 1.7:1.
It was worth noting that the pH value is the most important factor that governs the performance of photocatalytic H2 evolution. Figure 2c shows pH effect on the H2 evolution under the same concentrations of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst, MPA-CdTe QDs, chitosan and H2A. A maximal rate of H2 evolution was achieved at pH 4.5, whereas significant amounts of H2 were also observed at either lower or higher pH value. This pH-dependent effect should be related to the solubility of chitosan, the stability of MPA-CdTe QDs and the equilibrium of H2A=H++HA−. At a higher pH value, the lack of protonatable amine groups at C-2 position of the glucosamine residue54 decreases the solubility of chitosan and thus lowers the ability of chitosan to function as an environmental confinement. On the other hand, the protons in the solution with lower pH would suppress the equilibrium to generate enough sacrificial electron donor of HA− for H2 evolution45, and at the same time the MPA ligands would dissociate from the surface of CdTe QDs at a pH value of the solution lower than 4.0, resulting in precipitation and defects that could capture the excited electrons on the surface of the MPA-CdTe species55,56.
Considering all above experimental trials, we carried out the reaction under the optimized condition, that is, 10 ml methanol/water solution (1:3 v-v) containing [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (1.00 × 10−6 mol l−1), MPA-CdTe QDs (1.71 × 10−6 mol l−1), H2A (0.10 mol l−1) and chitosan (1.0 g l−1) at pH 4.5. More than 1.04±0.04 ml of H2 was produced during 10 h of irradiation with visible light (λ=410 nm) (Supplementary Fig. S4). Even more amounts of H2 in a total of 11.83±0.39 ml were obtained when the concentration of H2A was further increased to 0.20 mol l−1 (Supplementary Fig. S6). This result means that more than (5.28±0.17) × 104 equivalents of H2 per [Fe2(CO)6(μ-adt)CH2C6H5] catalyst are generated over 60 h of irradiation (Fig. 2d), with an initial TOF of 1.40±0.22 H2 per catalyst per second in the first 2 h (Supplementary Fig. S7). The catalytic activity is improved 4.16 × 103 folds that of the same system without chitosan.
Interaction of catalytic components with chitosan
The enhanced durability and efficiency is possibly due to the strong interaction and close contact between the MPA-CdTe QDs, [Fe2(CO)6(μ-adt)CH2C6H5] catalyst and H2A in the chitosan-confined environment (Fig. 1). The encapsulation of the MPA-CdTe QDs by chitosan was well evidenced by high-resolution transmission electron microscopy. The high-resolution transmission electron microscopy images of the MPA-CdTe QDs reveal that chitosan associates with the MPA-CdTe QDs to form self-assemblies on a large scale, and their average size is in the range of 50~200 nm (Fig. 3). Even after 10 h of irradiation, the shape and composition of the self-assemblies with well-crystallized lattices of MPA-CdTe QDs for H2 evolution remained unchanged. This finding is different from that observed in the reaction system without chitosan (Supplementary Fig. S8). Although no obvious spectral change could be detected in the UV–vis absorption spectra of chitosan and MPA-CdTe QDs as well as their mixture (Supplementary Fig. S9), the photoluminescent intensity of the MPA-CdTe QDs increased and blue-shifted greatly with the introduction of chitosan (Fig. 4a), and simultaneously the photoluminescent lifetime of the MPA-CdTe QDs enhanced from 10.9 to 18.3 ns when the concentration of chitosan was varied from 0 to 1.0 g l−1 at pH 4.5 (Fig. 4b). It is known that the photoluminescence of QDs is very sensitive to a pH value of solution55. When the pH value of an aqueous solution of the MPA-CdTe was adjusted to 4.5, the maximal photoluminescence was found to shift to lower energy at 575 nm accompanying with decreases in the photoluminescent intensity and lifetime (Table 1). The observations are due to the aggregation of the MPA-CdTe QDs to form larger ones55,56. The blue-shift from 575 to 557 nm in the current study suggests that chitosan stabilizes the CdTe QDs and prevents them from forming larger aggregators. More strikingly, the photoluminescence quantum yield of the MPA-CdTe QDs increased from 5.1% to 38.3% when chitosan was presented in the solution of methanol/water (1:3, v-v) at pH 4.5. The photoluminescent enhancement in intensity, lifetime and quantum yield indicates that chitosan wraps the MPA-CdTe QDs by coordination to cadmium ions of CdTe QDs, and thus suppresses, to some extent, the non-radiative decay of MPA-CdTe QDs. The similar effect was also observed by Yang and Gao et al.57 with the addition of poly(acrylic acid) into the aqueous solution of CdTe QDs.

High-resolution transmission electron microscopy images of MPA-CdTe QDs and chitosan in the reaction system before irradiation (a) (bar scale, 200 nm) and after irradiation for 10 h (b) (bar scale, 200 nm). The MPA-CdTe QDs inside chitosan after irradiation for 10 h (c) (bar scale, 2 nm).

(a) Photoluminescence spectra of MPA-CdTe QDs (0.86 × 10−6 mol l−1) at pH 10 and pH 4.5, and the photoluminescence of MPA-CdTe in the presence of chitosan at pH 4.5, in methanol/water (1:3 v-v). (b) Photoluminescence lifetime of MPA-CdTe QDs in the absence and presence of chitosan (1.0 g l−1) at pH 4.5. The signal of IRF (blue) is the response of the instrument.
The interaction of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst with chitosan was carefully examined and is shown in Fig. 5. No absorbance could be detected from [Fe2(CO)6(μ-adt)CH2C6H5] catalyst in pure water but with continuous sonication of insoluble [Fe2(CO)6(μ-adt)CH2C6H5] catalyst and a chitosan (1.0 g l−1) solution in methanol/water (1:3, v-v) at pH 4.5 its solubility and absorbance were remarkably enhanced with the formation of a coloured solution. Alternatively, progressive addition of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst in dichloromethane into a solution of chitosan (1.0 g l−1) in methanol/water (1:3, v-v) at pH 4.5 resulted in an increase of absorption band at 336 nm remarkably. As compared with the same system without chitosan in methanol/water (1:3, v-v) at pH 4.5 (Supplementary Fig. S10), the increment of the absorbance at 336 nm is much greater. The results indicate that water-insoluble [Fe2(CO)6(μ-adt)CH2C6H5] catalyst is incorporated into the chitosan solution. The absorbance at 336 nm obeys the Beer’s law showing that [Fe2(CO)6(μ-adt)CH2C6H5] catalyst is well dispersed in the chitosan solution at pH 4.5. Decreasing the pH of the solution has no noticeable influence on the absorption spectra, suggesting that [Fe2(CO)6(μ-adt)CH2C6H5] catalyst does not react with protons under the experimental condition.

(a) The absorption spectra of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (3.00 × 10−7 mol) in water at pH 4.5, an chitosan solution (1.0 g l−1) in methanol/water (1:3, v-v) at pH 4.5 and [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (3.00 × 10−7 mol) with continuous sonication of the chitosan solution (1.0 g l−1) in methanol/water (1:3, v-v) at pH 4.5. (b) The absorption spectra of chitosan (1.0 g l−1) with progressive addition of [Fe2(CO)6(μ-adt)CH2C6H5] (1.00 × 10−3 mol l−1 in CH2Cl2) in methanol/water (1:3, v-v) at pH 4.5; inset: the absorbance at 336 nm as a function of amounts of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst. (c) The schematic representation of dialysis experiment: the 10 ml solution inside the dialyser containing [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (1.00 × 10−4 mol l−1) in the absence (A) and presence (B) of chitosan (1.0 g l−1) in methanol/water (1:3, v-v) at pH 4.5; the solution outside the dialyser containing 90 ml of methanol/water (1:3, v-v) at pH 4.5. (d) The time course of the concentration changes of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst outside the solution of dialysis bag A and dialysis bag B, respectively, which was read from UV–vis absorption spectra. (e) Cyclic voltammograms of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (2.0 × 10−4 mol l−1) in the absence and presence of HOAc in methanol/water (1:1, v-v). (f) Cyclic voltammograms of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (2.0 × 10−4 mol l−1) in the absence and presence of HOAc in methanol/water (1:1, v-v) with chitosan (1.0 g l−1).
The interaction of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst and chitosan was further confirmed by dialysis experiments. As depicted in the schematic representation of Fig. 5, 1.00 × 10−4 mol l−1 of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst was put into dialysis bag A, and the same amount of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst together with 1.0 g l−1 of chitosan were in dialysis bag B, respectively. Along with the time, the diffusion rate of the catalyst to the outside solution from dialysis bag B was noted much slower than that from dialysis bag A, and thus leading to the concentration change of the catalyst from dialysis bag B smaller than that from dialysis bag A. These results imply the intimate interaction of chitosan and [Fe2(CO)6(μ-adt)CH2C6H5] catalyst.
The direct evidence on the interaction comes from electrochemical studies under nitrogen atmosphere. Note that the reduction potential of [Fe2(CO)6(μ-adt)CH2C6H5] positively shifts from −1.36 V versus NHE in acetonitrile to −1.10 V versus NHE in methanol/water (1:1, v-v) at pH 4.5 (Supplementary Fig. S11), which is attributed to the reduction of FeIFeI to FeIFe0 of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst27,28,29. Although the reduction potential of [Fe2(CO)6(μ-adt)CH2C6H5] remained unchanged with the addition of chitosan at pH 4.5 (Table 1), the cyclic voltammogram of a solution with or without chitosan, containing the same amounts of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst, displayed different electrochemical responses on progressive addition of acetic acid (HOAc). Given in Fig. 5 is the cyclic voltammetry of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst in the absence and presence of HOAc. The current intensity of the reduction peak increases with the acid concentration, the characteristic of proton reduction10,11,12. On reversing the scan following the reductions at −1.26 V versus NHE, a reproducible curve-crossing was clearly observed for [Fe2(CO)6(μ-adt)CH2C6H5] catalyst resulting in the buildup of current response at −1.00 V versus NHE. The peak current at −1.00 V is proportional to the square root of the scan rate (Supplementary Fig. S12), suggesting that the electrochemical processes are diffusion-controlled and excluding the possibility of the curve-crossing event arising from electrode deposition. Moreover, the current height of the −1.00 V events increases with increasing acid concentrations. Its dependence on both potential scan rate and acid concentration reveals that a larger fraction of the starting material is regenerated at reaction times that correspond to potentials positive of the curve-crossing. Following from the electrochemical studies of a mimic of the diiron subunit of [FeFe]-H2ase by Darensbourg and co-workers28, we suppose that the curve-crossing electrochemical responses are an integral property of the electroactive (−1.10 V) species, presumed to be the FeIFeI to FeIFe0 reduction, for which a rapid chemical reaction, that is, protonation of the FeIFe0 species produces the increased current at more negative potential. The presence of a more easily reproducible product or intermediate as seen in the reverse electrochemical scan suggests that a subsequent slow chemical reaction produces an intermediate of sufficient stability to build up in solution and migrate back to the electrode for reduction at a more positive potential. Evidently, the system with chitosan yielded much more intermediate species at −1.00 V than that working in the absence of chitosan under the same concentration of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst, indicative of greater sensitivity and stability of the reduced species to acid concentration in the presence of chitosan.
In view of the sensitivity of the system to the pH value of solution, all of the interaction studies were carried out at pH 4.5 to agree with the optimized condition. In the presence of chitosan, the absorption spectrum of the MPA-CdTe QDs and [Fe2(CO)6(μ-adt)CH2C6H5] catalyst was the superposition of the MPA-CdTe QDs, chitosan and [Fe2(CO)6(μ-adt)CH2C6H5] (Supplementary Fig. S9), but the photoluminescence of MPA-CdTe QDs was quenched by [Fe2(CO)6(μ-adt)CH2C6H5] dramatically. As shown in Fig. 6, excitation of the characteristic absorption of MPA-CdTe QDs resulted in a maximal photoluminescence at 575 nm in methanol/water (1:3, v-v) solution, which was quenched by [Fe2(CO)6(μ-adt)CH2C6H5] with a rate constant of (9.95±0.67) × 103 l mol−1 (Table 1, Supplementary Fig. S13). When 1.0 g l−1 of chitosan was presented in the solution, the photoluminescent maximum blue-shifted to 557 nm and the quenching rate constant increased to (2.26±0.02) × 104 l mol−1 (Table 1, Supplementary Fig. S13). Clearly, the interaction between the MPA-CdTe QDs and [Fe2(CO)6(μ-adt)CH2C6H5] catalyst is stronger in the self-assembled chitosan system than that in free solution.

(a) Photoluminescence quenching of MPA-CdTe QDs (0.86 × 10−6 mol l−1) with progressive addition of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst in the absence of chitosan. (b) Photoluminescence quenching of MPA-CdTe QDs (0.86 × 10−6 mol l−1) with progressive addition of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst in the presence of chitosan. (c) Transient absorption spectra of MPA-CdTe QDs (0.86 × 10−6 mol l−1) and [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (5.00 × 10−5 mol l−1) in the absence (top) and presence (bottom) of chitosan in methanol/water (1:3 v-v) at pH 4.5.
As the spectral overlap of absorption of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst and photoluminescence of MPA-CdTe QDs is rather small, the energy transfer from the excited MPA-CdTe QDs to [Fe2(CO)6(μ-adt)CH2C6H5] catalyst would be negligible. Electron transfer from the excited MPA-CdTe QDs to [Fe2(CO)6(μ-adt)CH2C6H5] catalyst is therefore responsible for the photoluminescence quenching of the MPA-CdTe QDs. Combining electrochemical and spectroscopic studies (Table 1), we estimated the free-energy change of electron transfer reaction from the excited MPA-CdTe QDs to [Fe2(CO)6(μ-adt)CH2C6H5] catalyst. According to the valence-band energy level (Evb) of MPA-CdTe QDs, which is 0.09 V (all potentials discussed here are versus NHE)58 and the reduction potential (Ered) of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst determined as −1.10 V in methanol/water (1:1, v-v) at pH 4.5 (Table 1), the excited-state energy (E00) of MPA-CdTe QDs being 2.23 eV in the presence of chitosan and 2.16 eV in the absence of chitosan at pH 4.5 (Table 1), respectively, the free-energy change (ΔG0) of the electron transfer reaction was thus calculated to be −1.04 eV in the presence of chitosan and −0.97 eV in the absence of chitosan at pH 4.5 (Table 1, Supplementary Fig. S14). This means that the electron transfer from the excited MPA-CdTe QDs to [Fe2(CO)6(μ-adt)CH2C6H5] catalyst in this designed system is more exothermic.
Flash photolysis study provides direct evidence on the photoinduced electron transfer process at room temperature. On laser excitation of the MPA-CdTe QDs using 355 nm light, no characteristic absorption signal was observed from ultraviolet to visible region under the time scale of 2.0 μs (Supplementary Fig. S15). When [Fe2(CO)6(μ-adt)CH2C6H5] catalyst was added into the MPA-CdTe QDs solution containing 1.0 g l−1 of chitosan, a new set of absorption bands emerged immediately (Fig. 6c). The generated new absorption is similar to that of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst under reduction potential at −1.16 V versus NHE in methanol/water (1:1, v-v) at pH 4.5 (Supplementary Fig. S15), in line with the FeIFe0 species reported by Pickett and co-workers29 using the same approach. Therefore, the absorption ~410 nm is attributed to the FeIFe0 species generated by electron transfer from the excited MPA-CdTe QDs to [Fe2(CO)6(μ-adt)CH2C6H5]. As that of the MPA-CdTe QDs after delivering an electron to [Fe2(CO)6(μ-adt)CH2C6H5] catalyst might show absorptions in this region though no signal was detected on laser excitation of MPA-CdTe QDs itself in methanol/water (1:3, v-v) at pH 4.5, we proposed that the transient signals at ~410 nm may result from the spectral overlap of both FeIFe0 species and that of the CdTe QDs after electron transfer. The active FeIFe0 species from [Fe2(CO)6(μ-adt)CH2C6H5] catalyst can further react with protons to experience catalytic cycle for H2 evolution. The formed hole remaining in the MPA-CdTe species after electron transfer, on the other hand, is subsequently regenerated by electron transfer from the sacrificial electron donor. It is known that the redox potential of H2A (−0.14 V at pH 4.5)59 is sufficiently negative to reduce the holes photogenerated in MPA-CdTe species25,45, but it is too positive to directly reduce the [FeFe]-H2ase catalyst (Supplementary Fig. S14). Therefore, the holes left in CdTe QDs are consumed by the sacrificial electron donor H2A.
To examine the possibility of chitosan to function as another electron donor, we did the experiment for H2 evolution in the absence of H2A at pH 4.5 (Supplementary Fig. S2). But no H2 could be detected in the system, indicative that the functionality of chitosan to serve as a sacrificial electron donor would be negligible. In this case, one may speculate that when the MPA-CdTe QDs is excited by visible light, the electron transfer from the conduction band of MPA-CdTe QDs to [Fe2(CO)6(μ-adt)CH2C6H5] catalyst takes place giving rise to the reduced [Fe2(CO)6(μ-adt)CH2C6H5]. At the same time, the holes remaining in the valence band of MPA-CdTe QDs after electron transfer are regenerated by electron transfer from the sacrificial ascorbic acid H2A to complete the photocatalytic cycle (Supplementary Fig. S14).
Discussion
As compared with those reported in the literature34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51, the durability and activity of the present system are greatly increased; possibly as a result of the stabilization of the components by chitosan confinement leading to consecutive multi-step electron transfer in equilibrium. The importance of the stabilization was also analysed by exchanging chitosan for relatively small and loose aggregates, anionic SDS (0.166 mol l−1) and cationic CTAB (0.055 mol l−1, cetyl trimethyl ammonium bromide) micelles37. For systematic comparison, the photocatalytic H2 evolution experiment was carried out from the same reaction system, containing MPA-CdTe QDs, [Fe2(CO)6(μ-adt)CH2C6H5] catalyst and H2A, in SDS and CTAB micelles, respectively. The TON is only 37.6±3.2 and 36.0±4.4 for the SDS and CTAB system, respectively, even after irradiation for 10 h (Supplementary Fig. S16). Notably, these solutions were quickly changed from orange to brownish red with the formation of brown precipitates on irradiation, whereas the chitosan-involved system was clear even after 40 h irradiation. The results demonstrate that the chitosan-confined H2-evolving system has more advantage over the micellar systems. The significant content of hydroxyl group and protonated amines of polycationic chitosan has shown affinity towards the negative MPA-CdTe QDs, [Fe2(CO)6(μ-adt)CH2C6H5] catalyst and H2A, which improves the electron transfer processes from the MPA-CdTe QDs to [Fe2(CO)6(μ-adt)CH2C6H5], as well as H2A to the holes of MPA-CdTe QDs after electron transfer (see above). Because two electrons are required to produce each molecule of H2, the stabilization of the MPA-CdTe QDs, [Fe2(CO)6(μ-adt)CH2C6H5] catalyst and catalytic intermediate and the consecutive multi-step electron transfer in equilibrium are believed to be responsible for the regeneration of the MPA-CdTe species and [Fe2(CO)6(μ-adt)CH2C6H5] catalyst to improve the efficiency of H2 evolution in the chitosan-confined system.
Unlike most state-of-the-art approaches, the system does not rely on further structure modification of butterfly [Fe2S2] subunit but on the addition of natural polysaccharide chitosan. The catalytic performance has been improved from 12.7±1.3 to (5.28±0.17) × 104 turnover numbers under the same condition, which increases 4.16 × 103 folds as compared with the same system without chitosan. These results imply that the environmental protein surrounding catalytic centre might cause the significant activity difference between the diiron subsite of natural [FeFe]-H2ase and its synthetic mimics. The crucial role of chitosan suggests that to create active H2 evolution systems based on artificial [FeFe]-H2ases, one would need to mimic not only the structure of active centre but also the biological environment surrounding [Fe2S2] subunit. The present artificial system using chitosan-confined environment is reminiscent of the [Fe2S2] subcluster of natural [FeFe]-H2ase buried in heterogeneous protein matrix, and demonstrates that artificial [FeFe]-H2ases are promising alternatives for use in a future sustainable H2 economy.
Methods
Chemicals and synthesis
All reagents were weighed and handled in air, and backfilled under an inert atmosphere of argon at room temperature. Chitosan (low molecular weight, 20–300 cP, 1 wt. % in 1% acetic acid (25 °C, Brookfield (lit.)), L-ascorbic acid (H2A, 99%), MPA (99%) and CdCl2·2.5H2O (99%) were purchased from Sigma-Aldrich. Benzylamine (97%) and paraformaldehyde (97%) were purchased from Alfa-Aesar. All commercial chemicals are used without further purification unless otherwise noted. The ultrapure water with 18.2 MΩ cm (Mettler Toledo, FE20) was used throughout the experiment.
The [Fe2(CO)6(μ-adt)CH2C6H5] catalyst was synthesized by the reaction of benzylamine, aldacide, thionylchloride and the lithium salt of diiron hexacarbonyldisulphide as that repoted in the previous work27,37. The aqueous colloidal MPA-CdTe QDs solution was prepared using the reaction between Cd2+ and NaHTe solution according to the literature45. Cd2+ precursor solutions were prepared by mixing the solutions of CdCl2·2.5H2O and stabilizer (MPA) followed by pH adjustment to 10 with 1 mol l−1 NaOH and degassed by bubbling nitrogen for 30 min. Then a fresh NaHTe was added under anaerobic condition in a typical molar ratio of Cd:MPA:Te as 1:1.2:0.2. The resulting solution was then heated to 99–100 °C after bubbling nitrogen for another 30 min and refluxed in different reaction time to control the size of MPA-CdTe QDs. Aliquots of the reaction solution were taken out at regular intervals for UV–vis absorption and photoluminescence characterization.
Photocatalytic H2 evolution
A typical procedure for H2 production is as follows. Chitosan (10 mg) and H2A (2.00 × 10−3 mol) were dissolved in 3.50 ml water and diluted with the 2.49 ml methanol with vigorous stirring. The excess acid was then neutralized by adding NaOH (5.0 mol l−1) solution and adjusted the solution to weakly acidic (pH 4~5). Then, 4.00 ml of aqueous MPA-CdTe QDs solution (1.71 × 10−6 mol l−1), 10.0 μl of methanol solution containing [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (1.00 × 10−3 mol l−1) were added to the above solution (total volume became 10 ml) with stirring. The pH value of the mixed solution was further adjusted to 4.5 by aqueous 1.0 mol l−1 HCl and determined by a pH meter. The sample was degassed by bubbling nitrogen for 30 min. Then 1,000 μl of CH4 was injected as the internal standard for quantitative GC analysis. The sample was irradiated by light-emitting diodes (λ=410 nm). The generated photoproduct of H2 was characterized by GC analysis (Tianmei 7890-II) using nitrogen as the carrier gas with a molecular sieve column (5 Å) and a thermal conductivity detector. Then 400 μl of mixed gas was extracted from the sample tube and injected into the GC immediately. The response factor for H2/CH4 was about 5.10 under the experimental condition, which was established by calibration with known amounts of H2 and CH4 and determined before and after a series of measurements. The desired concentration of reaction system was achieved by dissolving different amount of chitosan, H2A, the MPA-CdTe QDs and [Fe2(CO)6(μ-adt)CH2C6H5] catalyst into 10 ml of the mixed aqueous solution.
Absorption and photoluminescence measurements
UV–vis spectra were measured on a Shimadzu UV-1601PC spectrophotometer in a quartz cell with an optical path length of 1 cm. The interaction of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (0.30 × 10−6 mol, solid) and chitosan (1.0 g l−1, in methanol/water (1:3, v-v)) was sonicated for 15 min before measurement. Photoluminescence was recorded on a Hitachi F-4500 spectrofluorimeter at room temperature. Photoluminescence lifetime was measured on the Edinburgh FLSP920 with excitation at 405 nm. The photoluminescence quenching experiment was performed by progressive addition of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst (1.0 × 10−3 mol l−1, in methanol) into the solution at pH 4.5. The volume of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst added to the system is so small that the volume change is ignored in the determination of the concentration.
Dialysis experiments
The dialyser bags (purchased from Biotopped, molecular weight cutoff 3,500) were pretreated with hot water and kept in deionized water before use. Mixed solution (10 ml) containing [Fe2(CO)6(μ-adt)CH2C6H5] and chitosan was loaded into the dialyser bag. Then, the seal-off dialyser bag was soaked in 90 ml solution of methanol/water (1:3, v-v) in the dark. UV–vis absorption spectrometer was employed to examine the concentration of [Fe2(CO)6(μ-adt)CH2C6H5] outside dialysis bag.
Electrochemical and spectroelectrochemical measurements
A three-electrode system was used for the measurement and bulk electrolysis, with a 3-mm glass carbon working electrode, a platinum wire counter electrode and a non-aqueous Ag/AgNO3 reference electrode for organic solution or a saturated calomel electrode (SCE) reference electrode for aqueous solution. The working electrode was polished with a 0.05 μm alumina paste and sonicated for 15 min before use. The electrolyte solution (0.1 mol l−1 of n-Bu4NPF6 in acetonitrile for organic solution, 0.1 mol l−1 of Na2SO4 for methanol/water (1:1 v-v) solution) was purged with argon for 30 min before measurement. Spectroelectrochemical experiment was performed in a quartz cell with an optical path length of 1 cm. Indium tin oxide glass was used as a working electrode and a platinum wire electrode and a SCE reference electrode were served as the counter and reference electrodes, respectively. The electrolyte solution was purged with argon for 30 min before the absorption spectra were recorded on a Shimadzu UV-1601PC spectrometer. Spectroelectrochemical absorption spectrum was recorded along with time of electrochemical reduction of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst at −1.4 V relative to SCE (−1.16 V versus NHE) in methanol/water (1:1, v-v), the baseline of which refers to the absorption of [Fe2(CO)6(μ-adt)CH2C6H5] catalyst before reduction under the voltage.
Flash photolysis
The transient absorption spectroscopy was recorded on Edinburgh LP 920 at room temperature. A mixture of methanol/water (1:3 v-v) solution was degassed with nitrogen for 30 min before measurement. Excitation was provided using Nd:YAG laser (third harmonic, 10 ns) at 355 nm and the detector was a xenon lamp on the Edinburgh LP 920 apparatus.
Additional information
How to cite this article: Jian, J.-X. et al. Chitosan confinement enhances hydrogen photogeneration from a mimic of the diiron subsite of [FeFe]-hydrogenase. Nat. Commun. 4:2695 doi: 10.1038/ncomms3695 (2013).
References
Frey, M. Hydrogenases: hydrogen-activating enzymes. Chembiochem. 3, 153–160 (2002).
Knörzer, P. et al. Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases. J. Biol. Chem. 287, 1489–1499 (2012).
Nicolet, Y. & Fontecilla-Camps, J. C. Structure-function relationships in [FeFe]-hydrogenase active site maturation. J. Biol. Chem. 287, 13532–13540 (2012).
Fontecilla-Camps, J. C., Volbeda, A., Cavazza, C. & Nicolet, Y. Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem. Rev. 107, 4273–4303 (2007).
Adams, M. W. W. & Stiefel, E. I. Biological hydrogen production: not so elementary. Science 282, 1842–1843 (1998).
Stripp, S., Sanganas, O., Happe, T. & Haumann, M. The structure of the active site H-cluster of [FeFe] hydrogenase from the green algae Chlamydomonas reinhardtii studied by X-ray absorption spectroscopy. Biochem. 48, 5042–5049 (2009).
Woolerton, T. W., Sheard, S., Chaudhary, Y. S. & Armstrong, F. A. Enzymes and bio-inspired electrocatalysts in solar fuel devices. Energy Environ. Sci. 5, 7470–7490 (2012).
Lubitz, W., Reijerse, E. J. & Messinger, J. Solar water-splitting into H2 and O2: design principles of photosystem II and hydrogenases. Energy Environ. Sci. 1, 15–31 (2008).
Hambourger, M. et al. Biology and technology for photochemical fuel production. Chem. Soc. Rev. 38, 25–35 (2009).
Tard, C. & Pickett, C. J. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem. Rev. 109, 2245–2274 (2009).
Gloaguen, F. & Rauchfuss, T. B. Small molecule mimics of hydrogenases: hydrides and redox. Chem. Soc. Rev. 38, 100–108 (2009).
Capon, J.-F., Gloaguen, F., Pétillon, F. Y., Schollhammer, P. & Talarmin, J. Electron and proton transfers at diiron dithiolate sites relevant to the catalysis of proton reduction by the [FeFe]-hydrogenases. Coord. Chem. Rev. 253, 1476–1494 (2009).
Lomoth, R. & Ott, S. Introducing a dark reaction to photochemistry: photocatalytic hydrogen from [FeFe] hydrogenase active site model complexes. Dalton. Trans. 45, 9952–9959 (2009).
Magnuson, A. et al. Biomimetic and microbial approaches to solar fuel generation. Acc. Chem. Res. 42, 1899–1909 (2009).
Wang, M. & Sun, L. Hydrogen production by noble-metal-free molecular catalysts and related nanomaterials. ChemSusChem 3, 551–554 (2010).
Concepcion, J. J., House, R. J., Papanikolas, J. M. & Meyer, T. J. Chemical approaches to artificial photosynthesis. Proc. Natl Acad. Sci. USA 109, 15560–15564 (2012).
Han, Z.-J., Qiu, F., Eisenberg, R., Holland, P. L. & Krauss, T. D. Robust photogeneration of H2 in water using semiconductor nanocrystals and a nickel catalyst. Science 338, 1321–1324 (2012).
Nocera, D. G. The artificial leaf. Acc. Chem. Res. 45, 767–776 (2012).
Balzani, V., Credi, A. & Venturi, M. Photochemical conversion of solar energy. ChemSusChem 1, 26–58 (2008).
Andreiadis, E. S., Chavarot-Kerlidou, M., Fontecave, M. & Artero, V. Artificial photosynthesis: from molecular catalysts for light-driven water splitting to photoelectrochemical cells. Photochem. Photobiol. 87, 946–964 (2011).
Frischmann, P. D., Mahata, K. & Wurthner, F. Powering the future of molecular artificial photosynthesis with light-harvesting metallosupramolecular dye assemblies. Chem. Soc. Rev. 42, 1847–1870 (2013).
Wang, F. et al. Artificial photosynthetic systems based on [FeFe]-hydrogenase mimics: the road to high efficiency for light-driven hydrogen evolution. ACS Catal. 2, 407–416 (2012).
Greene, B. L., Joseph, C. A., Maroney, M. J. & Dyer, R. B. Direct evidence of active-site reduction and photodriven catalysis in sensitized hydrogenase assemblies. J. Am. Chem. Soc. 134, 11108–11111 (2012).
Reisner, E., Powell, D. J., Cavazza, C., Fontecilla-Camps, J. C. & Armstrong, F. A. Visible light-driven H2 production by hydrogenases attached to dye-sensitized TiO2 nanoparticles. J. Am. Chem. Soc. 131, 18457–18466 (2009).
Brown, K. A., Dayal, S., Ai, X., Rumbles, G. & King, P. W. Controlled assembly of hydrogenase-CdTe nanocrystal hybrids for solar hydrogen production. J. Am. Chem. Soc. 132, 9672–9680 (2010).
Gloaguen, F., Lawrence, J. D. & Rauchfuss, T. B. Biomimetic hydrogen evolution catalyzed by an iron carbonyl thiolate. J. Am. Chem. Soc. 123, 9476–9477 (2001).
Barton, B. E., Olsen, M. T. & Rauchfuss, T. B. Aza- and oxadi-thiolates are probable proton relays in functional models for the [FeFe]-hydrogenases. J. Am. Chem. Soc. 130, 16834–16835 (2008).
Mejia-Rodriguez, R., Chong, D., Reibenspies, J. H., Soriaga, M. P. & Darensbourg, M. Y. The hydrophilic phosphatriazaadamantane ligand in the development of H2 production electrocatalysts: Iron hydrogenase model complexes. J. Am. Chem. Soc. 126, 12004–12014 (2004).
Borg, S. J. et al. Electron-transfer at a dithiolate-bridged di-Iron assembly; electrocatalytic hydrogen evolution. J. Am. Chem. Soc. 126, 16988–16999 (2004).
Quentel, F., Passard, G. & Gloaguen, F. Electrochemical hydrogen production in aqueous micellar solution by a diiron benzenedithiolate complex relevant to [FeFe] hydrogenases. Energy Environ. Sci. 5, 7757–7761 (2012).
Camara, J. M. & Rauchfuss, T. B. Combining acid–base, redox and substrate binding functionalities to give a complete model for the [FeFe]-hydrogenase. Nat. Chem. 4, 26–30 (2012).
Jones, A. K., Lichtenstein, B. R., Dutta, A., Gordon, G. & Dutton, P. L. Synthetic hydrogenases:incorporation of an iron carbonyl thiolate into a designed peptide. J. Am. Chem. Soc. 129, 14844–14845 (2007).
Singleton, M. L., Reibenspies, J. H. & Darensbourg, M. Y. A cyclodextrin host/guest approach to a hydrogenase active site biomimetic cavity. J. Am. Chem. Soc. 132, 8870–8871 (2010).
Ott, S., Kritikos, M., Åkermark, B. & Sun, L. Synthesis and structure of a biomimetic model of the iron hydrogenase active site covalently linked to a ruthenium photosensitizer. Angew. Chem. Int. Ed. 42, 3285–3288 (2003).
Na, Y. et al. Visible light-driven electron transfer and hydrogen generation catalyzed by bioinspired [2Fe2S] complexes. Inorg. Chem. 47, 2805–2810 (2008).
Streich, D. et al. High-turnover photochemical hydrogen production catalyzed by a model complex of the [FeFe]-hydrogenase active site. Chem. Eur. J. 16, 60–63 (2010).
Wang, H.-Y. et al. Photocatalytic hydrogen evolution from rhenium(I) complexes to [FeFe] hydrogenase mimics in aqueous SDS micellar systems: a biomimetic pathway. Langmuir. 26, 9766–9771 (2010).
Kluwer, A. M. et al. Self-assembled biomimetic [2Fe2S]-hydrogenase-based photocatalyst for molecular hydrogen evolution. Proc. Natl Acad. Sci. USA 106, 10460–10465 (2009).
Wang, W.-G. et al. Photocatalytic hydrogen evolution by [FeFe] hydrogenase mimics in homogeneous solution. Chem. Asian J. 5, 1796–1803 (2010).
Samuel, A. P. S., Co, D. T., Stern, C. L. & Wasielewski, M. R. Ultrafast photodriven intramolecular electron transfer from a zinc porphyrin to a readily reduced diiron hydrogenase model complex. J. Am. Chem. Soc. 132, 8813–8815 (2010).
Wang, W.-G., Wang, F., Wang, H.-Y., Tung, C.-H. & Wu, L.-Z. Electron transfer and hydrogen generation from a molecular dyad: platinum(II) alkynyl complex anchored to [FeFe] hydrogenase subsite mimic. Dalton Trans. 41, 2420–2426 (2012).
Poddutoori, P. et al. Photoinitiated multistep charge separation in ferrocene-zinc porphyrin-diiron hydrogenase model complex triads. Energy Environ. Sci. 4, 2441–2450 (2011).
Wang, H.-Y. et al. A triad [FeFe] hydrogenase system for light-driven hydrogen evolution. Chem. Commun. 47, 8406–8408 (2011).
Nann, T. et al. Water splitting by visible light: a nanophotocathode for hydrogen production. Angew. Chem. Int. Ed. 49, 1574–1577 (2010).
Wang, F. et al. A highly efficient photocatalytic system for hydrogen production by a robust hydrogenase mimic in an aqueous solution. Angew. Chem. Int. Ed. 50, 3193–3197 (2011).
Wen, F. et al. A hybrid photocatalytic system comprising ZnS as light harvester and an [Fe2S2] hydrogenase mimic as hydrogen evolution catalyst. ChemSusChem 5, 849–853 (2012).
Cao, W.-N. et al. Photocatalytic hydrogen production from a simple water-soluble [FeFe]-hydrogenase model system. Chem. Commun. 48, 8081–8083 (2012).
Roy, A., Madden, C. & Ghirlanda, G. Photo-induced hydrogen production in a helical peptide incorporating a [FeFe] hydrogenase active site mimic. Chem. Commun. 48, 9816–9818 (2012).
Sano, Y., Onoda, A. & Hayashi, T. A hydrogenase model system based on the sequence of cytochrome c: photochemical hydrogen evolution in aqueous media. Chem. Commun. 47, 8229–8231 (2011).
Li, X., Wang, M., Zheng, D., Han, K., Dong, J. & Sun, L. Photocatalytic H2 production in aqueous solution with host-guest inclusions formed by insertion of an FeFe-hydrogenase mimic and an organic dye into cyclodextrins. Energy Environ. Sci. 5, 8220–8224 (2012).
Wang, W.-G. & Rauchfuss, T. B. Unsensitized photochemical hydrogen production catalyzed by diiron hydrides. J. Am. Chem. Soc. 134, 4525–4528 (2012).
Wu, S., Zeng, F., Zhu, H. & Tong, Z. Energy and electron transfers in photosensitive chitosan. J. Am. Chem. Soc. 127, 2048–2049 (2005).
Yamada, Y., Hozumi, K. & Nomizu, M. Construction and activity of a synthetic basement membrane with active laminin peptides and polysaccharides. Chem. Eur. J. 17, 10500–10508 (2011).
Kiang, T., Wen, J., Lim, H. W. & Leong, K. W. The effect of the degree of chitosan deacetylation on the efficiency of gene transfection. Biomaterials 25, 5293–5301 (2004).
Aldana, J., Lavelle, N., Wang, Y. & Peng, X. Size-dependent dissociation pH of thiolate ligands from cadmium chalcogenide nanocrystals. J. Am. Chem. Soc. 127, 2496–2504 (2005).
Zhang, Y., Mi, L., Wang, P.-N., Ma, J. & Chen, J.-Y. pH-Dependent aggregation and photoluminescence behavior of thiol-capped CdTe quantum dots in aqueous solutions. J. Lumin. 128, 1948–1951 (2008).
Zhang, H., Zhou, Z. & Yang, B. The influence of carboxyl groups on the photoluminescence of mercaptocarboxylic acid-stabilized CdTe nanoparticles. J. Phys. Chem. B 107, 8–13 (2003).
Rajh, T., Mićić, O. I. & Nozik, A. J. Synthesis and characterization of surface-modified colloidal CdTe qunatum dots. J. Phys. Chem. 97, 11999–12003 (1993).
Borsook, H. & Keighley, G. Oxidation-reduction potential of ascorbic acid (vitamin C). Proc. Natl Acad. Sci. 19, 875–878 (1933).
Karstens, T. & Kobs, K. Rhodamine B and rhodamine 101 as reference substances for fluorescence quantum yiels measurements. J. Phys. Chem. 84, 1871–1872 (1980).
Acknowledgements
We are grateful for financial support from the Ministry of Science and Technology of China (2013CB834505, 2013CB834804 and 2014CB239402), the National Natural Science Foundation of China (91027041, 21090343 and 51373193), the Solar Energy Initiative of the Knowledge Innovation Program of Chinese Academy of Sciences and the Bureau for Basic Research of the Chinese Academy of Sciences. We also acknowledge Ms. Yuejuan Ma for her help with the preparation of graphics.
Author information
Authors and Affiliations
Contributions
L.-Z.W. designed the research and supervised the whole project. Q.L. initiated the exploration in experiments and contributed to data analysis. J.-X.J. and Q.L. prepared samples, and performed experiments with input from L.-Z.W. Z.-J.L., F.W., Q.-Y.M., K.F., B.C. and C.-H.T. helped with the discussion. X.-B.L. performed the high-resolution transmission electron microscopy measurements. B.L. helped in the electrochemical and spectroelectrochemical measurements. Z.-J.L. and C.-B.L. provided CdTe QDs and the [Fe2(CO)6(μ-adt)CH2C6H5] catalyst. L.-Z.W. and Q.L. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S16, Supplementary Table S1 and Supplementary References (PDF 1358 kb)
Rights and permissions
About this article
Cite this article
Jian, JX., Liu, Q., Li, ZJ. et al. Chitosan confinement enhances hydrogen photogeneration from a mimic of the diiron subsite of [FeFe]-hydrogenase. Nat Commun 4, 2695 (2013). https://doi.org/10.1038/ncomms3695
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms3695
This article is cited by
-
Inspiring engineers
Communications Engineering (2022)
-
Photocatalysis-mediated drug-free sustainable cancer therapy using nanocatalyst
Nature Communications (2021)
-
Efficient photocatalytic hydrogen evolution with ligand engineered all-inorganic InP and InP/ZnS colloidal quantum dots
Nature Communications (2018)
-
Semiconducting quantum dots for artificial photosynthesis
Nature Reviews Chemistry (2018)
-
Efficient defect-controlled photocatalytic hydrogen generation based on near-infrared Cu-In-Zn-S quantum dots
Nano Research (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
