
Abstract
Real-time monitoring of nitric oxide concentrations is of central importance for probing the diverse roles of nitric oxide in neurotransmission, cardiovascular systems and immune responses. Here we report a new design of nitric oxide sensors based on hemin-functionalized graphene field-effect transistors. With its single atom thickness and the highest carrier mobility among all materials, graphene holds the promise for unprecedented sensitivity for molecular sensing. The non-covalent functionalization through π–π stacking interaction allows reliable immobilization of hemin molecules on graphene without damaging the graphene lattice to ensure the highly sensitive and specific detection of nitric oxide. Our studies demonstrate that the graphene–hemin sensors can respond rapidly to nitric oxide in physiological environments with a sub-nanomolar sensitivity. Furthermore, in vitro studies show that the graphene–hemin sensors can be used for the detection of nitric oxide released from macrophage cells and endothelial cells, demonstrating their practical functionality in complex biological systems.
Similar content being viewed by others


Introduction
Nitric oxide (NO) is believed to have diverse and vital roles in neurotransmission, cardiovascular systems and immune responses1,2. Since NO was first discovered as an elusive endothelial-derived relaxing factor in 1987, extensive research efforts have been devoted to studying the function of NO in physiology and pathology3,4,5. To comprehensively understand the physiological roles of NO and to design new strategies to manipulate NO regulatory pathways for therapeutic purposes, it is desirable to probe the production and diffusion processes of NO in biological systems in real time. To this end, the reliable and specific detection of NO with high spatiotemporal resolution is essential. However, the accurate determination of NO concentration is of significant challenge because of its low concentration (nanomolar scale) and relatively short half-life time (6–50 s) in biological systems6,7. Indirect detection methods—which rely on sensing secondary species such as nitrite and nitrate—are inherently ineffective for real-time detection. A number of direct detection methods such as electron paramagnetic resonance spectroscopy, chemiluminescence, mass spectrometry and fluorescence8,9,10 are being developed for NO detection in biological systems. However, most of these methods either suffer from low sensitivity or require complicated sample preparation or measurement conditions that preclude real-time detection7.
Electrochemical NO-sensing electrodes allow for real-time detection of NO with high sensitivity and selectivity11,12,13,14. However, as the signal amplitude and sensitivity of electrochemical sensors are generally proportional to the electroactive surface area of the electrodes13, it is often difficult to simultaneously achieve high sensitivity and high-spatial resolution. Electrochemical NO-sensing electrodes with low nanomolar detection limit usually require an active area of 1,500–30,000 μm2 (refs 7, 13, 14), and therefore cannot be used for high-spatial resolution measurements of NO distribution at a single-cell level, which is essential for the understanding of the signal transduction mechanisms of NO in physiology. In addition, electrochemical NO sensors usually operate at a relatively high potential (typically >0.6 V) for the electrochemical oxidation or reduction of NO to occur, which can disturb the intrinsic cellular response or induce significant signal interferences from other electrochemically active molecular species. The application of a coating of NO-selective gas permeable membrane on the electrode can improve the selectivity towards NO. However, greater selectivity usually requires thicker membrane, which not only complicates the fabrication process but also lowers the sensitivity and retards the response speed7.
With specific molecular functionalization, semiconductor (for example, GaAs)-based field-effect transistors (FETs) can be configured as NO sensors with reduced dimension for real-time detection of NO in physiological solutions15. However, the sensitivity of such sensors reported to date is typically limited to the micromolar regime, unsuitable for practical monitoring of biological NO levels that are typically in the nanomolar regime. Nanostructured materials are of increasing interest for the development of highly sensitive chemical and biological sensors16,17. For example, with the unique single atom thickness and exceptional electrical properties18,19,20,21,22,23,24,25, graphene has recently received considerable attention for this purpose26,27,28,29,30,31,32,33,34.
Here we report the construction of hemin-functionalized graphene FET as a new NO-sensing device for highly sensitive and selective detection of NO in physiological conditions. With the single atom thickness and the highest carrier mobility of all known materials, graphene-based NO sensors hold the promise for exceptional sensitivity that is not possible with other conventional materials for real-time, direct electrical read-out of NO signals. We show that the graphene–hemin sensors can rapidly and selectively respond to NO in physiological solutions with a sub-nanomolar sensitivity, and can be used for real-time monitoring of NO released from macrophage cells and endothelial cells (ECs), demonstrating their practical functionality in complex biological systems.
Results
Fabrication and characterization of graphene–hemin sensors
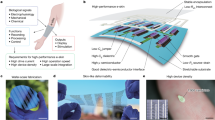
Graphene was prepared by mechanical exfoliation onto a silicon substrate with a 300-nm thermal oxide layer. Titanium-gold thin film source, drain and solution gate electrodes were fabricated using e-beam lithography, vacuum metal deposition, followed by a lift-off process. A typical graphene FET device has an active channel area ranging from 0.5 × 0.5 to 0.5 × 2 μm2, which is coupled with a polydimethylsiloxane (PDMS) microfluidic channel (with 0.5 mm channel height, 1 mm channel width and 10 mm channel length) for sample delivery (Fig. 1a). The graphene FET was non-covalently functionalized with hemin chloride through the π–π stacking interaction to form an NO sensor (Fig. 1b)35. Hemin is chosen as the functionalization molecule because of its large binding constant and high selectivity towards NO36. The non-covalent π–π interaction allows for the functionalization without damaging the graphene lattice or degrading its electronic performance.

(a) Schematic of a graphene sensor with integrated microfluidic sample delivery system. (b) Schematic illustration of a graphene–hemin FET that can selectively respond to NO. A typical graphene device has an active area of 0.5 × 0.5–0.5 × 2 μm2, sizes of source and drain electrodes are 2 × 5 μm2 (excluding the electrical leads) and the size of gate electrode is 50 × 50 μm2. The microfluidic PDMS channel has a width of 1 mm, height of 0.5 mm and length of 1 cm.
Ultraviolet–visible absorption spectroscopy is used to characterize the immobilization of hemin molecules on graphene (Fig. 2a). Before hemin functionalization, graphene on quartz only shows expected nearly flat absorption around 2.3% in the visible range (black line in Fig. 2a). After hemin molecules are conjugated with graphene, a clear absorption peak is observed at 425 nm (red line in Fig. 2a), close to the Soret band of hemin. The control sample with free hemin in dimethyl sulfoxide (DMSO) solution exhibits an absorption peak at 403 nm (blue line in Fig. 2a). The red shift of the Soret absorption peak can be attributed to the π–π interaction between graphene and hemin, which rotates the ethylene groups to the coplanar conformation with the porphyrin ring and flattens the hemin molecule to enhance the π conjugation37. We have quantified the amount of hemin molecules absorbed on graphene to be 0.94-hemin molecule per square nanometer, nearly a complete monolayer of hemin on graphene (see Supplementary Fig. S1 and Supplementary Note 1).

(a) Ultraviolet–visible spectrum of graphene before and after hemin immobilization. Black: bare graphene; red: graphene after immersed in hemin–DMSO solution, blue: hemin in DMSO solution. (b) AFM image of graphene after hemin immobilization. Scale bar, 1 μm. (c) Raman spectra of graphene before and after hemin functionalization. Black: bare graphene; blue: graphene after immersed in DMSO, with an offset of 0.2; red: graphene–hemin conjugate, with an offset of 0.4. (d) Real-time electrical measurement with NO precursor solution directly added onto a graphene–hemin device inside a PDMS well filled with PBS solution. Graphene–hemin device (red) and bare graphene device (black) were measured in parallel within the same PDMS well. Solution A is 0.01 M NaOH. B is NO precursor in PBS bubbled with N2 for 2 h (essentially the same as C but with NO removed). C is NO precursor in NaOH solution (both B and C are 1 μM after diluted in the PDMS well).
Atomic force microscope (AFM) is also used to characterize the hemin-functionalized graphene. To this end, e-beam lithography is used to open up a 3-μm-wide window, where graphene is selectively functionalized with hemin. AFM image of the selectively functionalized graphene shows that the functionalized area exhibits a clear height difference of ~0.5 nm from the unfunctionalized area (Fig. 2b), which can be attributed to the formation of a monolayer of hemin molecules on graphene.
Raman spectroscopy is used to further characterize the immobilization of hemin molecules on graphene (Fig. 2c). The Raman spectrum of bare graphene (black line) shows two main peaks: the G band (1,582 cm−1) and the 2D band (2,700 cm−1), characteristics of graphene38. After hemin functionalization, several new bands are observed at 1,370 cm−1, 1,631 cm−1, 1,430 cm−1 and 1,307 cm−1 (red line), which can be assigned for ν4, ν10, ν28 and ν21 vibrational modes of hemin molecules, respectively39. A decrease of the 2D band intensity is also observed after hemin immobilization. It was reported that the intensity of the 2D band decreases as a result of the interaction between graphene with either electron-donating or electron-accepting molecules40. As a control experiment, a graphene sample is processed in DMSO (the solvent) without hemin through the same procedures of functionalization (blue line). No apparent change is observed in the Raman spectrum compared with the bare graphene before processing, further supporting that the new Raman features (red line) observed in the hemin–graphene conjugate indeed originate from the immobilized hemin on graphene rather than any other processing-induced effects. Hence, these studies clearly demonstrate that hemin molecules are successfully immobilized on graphene.
Real-time electrical detection of NO
We then tested the electrical responses of graphene devices to NO solutions in real time. All the following real-time electrical measurements were conducted at a constant bias voltage of 10 mV and solution gate of 0 V, unless otherwise stated. The leakage current through the solution gate is typically <3 nA (Supplementary Fig. S2), which is negligible compared with the typical source drain current of the graphene–hemin devices (~1 μA). Two devices on the same substrate were kept in a PDMS well filled with 10 mM phosphate buffered saline (PBS) (pH=7.4), with only one of the devices selectively functionalized with hemin (set-up as in Supplementary Fig. S3). Both devices exhibited positive Dirac points with p-type characteristics at 0 V solution gate voltage bias (hole-transport branch). We used diethylamine NONOate sodium as a precursor to release NO in PBS (pH=7.4)41,42,43. At neutral pH, each one of these precursor molecules can hydrolyse to generate two molecules of NO, following a first-order reaction kinetics with a half-life time of ~2 min (ref. 41). At basic pH (pH≥12), this hydrolysis reaction proceeds much more slowly and is nearly negligible compared with that at neutral pH. Two devices were tested in parallel. With the introduction of 0.2 μl of a 10 mM NaOH aqueous solution (Fig. 2d, point A) or 0.2 μl of the precursor/PBS solution, in which NO had been eliminated by 2 h of N2 bubbling (Fig. 2d, point B), neither devices showed any obvious conductance change. In contrast, upon the introduction of 0.2 μl of a 100 μM diethylamine NONOate sodium in NaOH solution into the PBS in the PDMS well (Fig. 2d, point C), the hydrolysis generation of NO is initiated at neutral pH value; the conductance of hemin-functionalized device exhibits a noticeable gradual drop (Fig. 2d, red line). The time-dependent conductance response curve gives a half-life time of ~135 s, consistent with the hydrolysis kinetics expected for the NO precursor at pH 7.4 (ref. 41). On the other hand, the unfunctionalized device does not show any obvious conductance change because there is no active sites for NO binding (Fig. 2d, black line). This study clearly demonstrates that hemin-functionalized graphene device can indeed respond specifically to NO in PBS solutions.
We have further determined the sensitivity of the graphene–hemin NO sensors. To this end, the NO-sensing experiments were performed using a microfluidic system with a PDMS channel integrated on top of the graphene device (Fig. 1a), with which the freshly prepared NO solutions of increasing concentrations were introduced into the PDMS channel through a syringe pump. Figure 3a shows a real-time electrical read-out of different concentrations of NO from 0 to 1,000 nM. The average response time of the steps (t90%) is about 2 s, largely limited by the diffusion at the interface of different concentrations of NO sample solutions (see Supplementary Note 2). The inherent response time of the sensor is likely to be much faster. To obtain the calibration curve for NO detection, the amount of the conductance change was plotted against the NO concentration (Fig. 3b). The calibration curve in Fig. 3b can be fitted with a logarithmic plot. At low NO background concentration (for example, <10 nM), a lowest absolute detection limit of 0.3 nM can be achieved with a signal-to-noise ratio of 3. With increasing background NO concentration, the absolute sensitivity is reduced. Nonetheless, a relative sensitivity of 3–10% (the percentage of the concentration variation over the background concentration) is maintained consistently even at the highest NO concentration owing to the nature of logarithmic relationship. As the conductance change of the sensor originates from the doping effect, it is expected that the sensor devices with higher transconductance should exhibit higher sensitivity. Indeed, the detection limits of different devices show a consistent trend with a lower detection limit achieved in the device with higher transconductance (Fig. 3c). Altogether, these studies clearly demonstrate that the graphene–hemin devices can function as highly sensitive sensors for the detection of NO molecules in PBS.

(a) Real-time electrical measurement of different concentrations of NO. (b) Calibration curve: conductance change (ΔG) versus NO concentration (CNO). The red line is the fitted curve in natural logarithmic scale. (c) Relationship of the detection limit versus the transconductance of different sensor devices. (d) Solution gate-dependent measurement at different concentrations of NO. Vg, solution gate voltage. (e) Raman spectra of graphene–hemin in NO solutions at different concentrations. Black: PBS; red: 100 nM NO, with 0.1 offset; blue: 1 μM NO, with 0.2 offset; violet: 10 μM NO, with 0.3 offset; green: 100 μM NO, with 0.4 offset.
To better understand the effect of NO on the electrical properties of the sensor device, the conductance of the device was plotted against solution gate voltage in PBS with different NO concentrations (Fig. 3d). The hemin-immobilized graphene devices show a consistent negative shift of the Dirac point with increasing NO concentration from 0 to 1,000 nM. This negative shift of the Dirac point can be attributed to the doping effect of the hemin complex to the graphene channel upon the binding of NO to the Fe(III) centre. Specifically, with the immobilization of hemin onto graphene, the Fe(III) centre in hemin can interact with graphene in a metal–ligand model, similar to the Fe(III) benzene complexes44. In this case, graphene donates π-electrons to the d-orbital of Fe(III) through a cation–π interaction. When NO molecules come to coordinate with and donate electrons to Fe(III) centres, those π-electrons are partially transferred back to the valence band of graphene because of the trans-effect of the ligands45. Therefore, the binding of NO to Fe(III) centres is equivalent to partial electron-doping to hemin-functionalized graphene. This electron-doping effect explains the negative shifting of the Dirac point upon the introduction of NO, and is consistent with the reduction of conductance in the hole-transport branch of the graphene devices. Importantly, when monitoring the graphene–hemin device in electron-transport branch, a conductance increase is observed upon the introduction of NO (Supplementary Fig. S4), consistent with the electron-donating nature of the NO-binding events.
Raman spectroscopic study was also used to characterize the graphene–hemin device in the PBS solution of NO (Fig. 3e). The G band is found to shift to lower frequencies when the NO concentration is increased, consistent with n-type doping to graphene by electron-donating molecules reported in literature40. On the other hand, the G band of bare graphene does not exhibit any significant shift of frequency with the increasing NO concentration (Supplementary Fig. S5). It is also important to note that the Raman shift is rather small and only noticeable at relatively high NO concentrations (>100 nM), whereas the electrical signal is detectable at a much lower concentration of NO down to ≤1 nM.
Selectivity and functionality in physiological solutions
We have further investigated the selectivity and specificity of the graphene–hemin devices as NO sensors. With the introduction of a series of potentially interfering chemicals including oxygen, hydrogen peroxide, sodium nitrate, sodium nitrite, L-citrulline, L-arginine and diethylamine NONOate sodium bubbled with N2 (which contains only the hydrolysed precursor but not NO), no obvious conductance change is observed (Fig. 4a). In contrast, a conductance drop is clearly observed with the addition of 1 nM NO, demonstrating the excellent selectivity of our graphene–hemin NO sensors. We have also tested additional potentially interfering chemicals including L-glutathione, sodium ascorbate, carbon monoxide, hydrogen sulphide, sodium thiocyanate and sodium cyanide with the same devices (Fig. 4b). At the same concentration of 100 nM, the device responds to NO with the strongest signal, demonstrating excellent selectivity of our device to NO.

(a) Selectivity measurement with the addition of a series of control solutions (1 μM) followed by NO solutions (1 nM and 10 nM). (b) Selectivity measurement with NO and potentially interfering chemicals (all chemicals have a concentration of 100 nM. GSH, L-glutathione; Asc, sodium ascorbate). Signal is defined by ΔG/G, where ΔG is the conductance change and G is the conductance. The signals are the averages of three devices. (c) Real-time electrical measurement at different concentrations of NO in DMEM. (d) Real-time electrical measurement at different concentrations of NO in fetal bovine serum.
To further demonstrate the practical functionality of the sensor in physiological solutions, we have also conducted the NO-sensing measurements in Dulbecco's modified eagle medium (DMEM). The DMEM solution is a typical cell culture medium, containing various kinds of proteins, amino acids, vitamins, inorganic salts and other compounds including glucose. Furthermore, we have also tested the sensor performance in real biological samples such as fetal bovine serum that contains a large variety of potentially interfering species at physiological levels. In both cases, stepwise responses are clearly observed from the NO sensor when different NO concentrations are introduced (Fig. 4c,d). Importantly, the detection limits of 0.4 and 1.4 nM were achieved in DMEM solution (see calibration curve shown in Supplementary Fig. S6a) and fetal bovine serum (see calibration curve shown in Supplementary Fig. S6b), respectively, demonstrating the proper functionality of our sensors in physiological solutions.
We have also evaluated the stability of the graphene–hemin sensors by measuring the sensitivity of the same device after 1 and 3 weeks of storage under ambient conditions (Supplementary Fig. S7). The sensitivity of the sensor is 54 μS per 1,000 nM originally, and became 53 μS per 1,000 nM (98%) after 1 week and 47 μS per 1,000 nM (89%) after 3 weeks, which are comparable to the electrochemical sensors7,12. This signal drift is probably because of nonspecific surface adsorptions, and the stability of the device may be further improved through storage in a controlled environment.
Real-time monitoring of NO released by living cells
Going a step further, we have also conducted in vitro studies to test the performance of the graphene–hemin NO sensors for real-time detection of extracellular NO generated from RAW 264.7 macrophage cells. It is well known that macrophage cells can generate NO with expression of inducible NO synthase (iNOS) when stimulated by lipopolysaccharide (LPS)46,47,48,49. RAW 264.7 cells were seeded at 5 × 105 cells per ml (cell suspension) onto the graphene–hemin device on the SiO2 substrate (inset, Fig. 5b) within a PDMS well (set-up as in Supplementary Fig. S3). The cells were cultured for 12 h at 37 °C in a 5% CO2 incubator before NO-sensing measurement. The sensing measurement was carried out at 37 °C.

(a,b) Real-time monitoring of NO released from Raw 264.7 macrophage cells. (a) Real-time measurement of the conductance of the sensor device for NO detection. (b) Real-time measurement of the NO concentration. Inset: optical microscope image of RAW 264.7 cells seeded on the graphene–hemin device. In both (a,b), (A) 100 ng ml−1 LPS, (B) 100 ng ml−1 LPS and 100 μM aminoguanidine (AG), (C) without LPS. (c,d) Real-time monitoring of NO released from HUVECs. (c) Real-time measurement of the conductance of the sensor device. (d) Real-time measurement of the NO concentration. Inset: optical microscope image of HUVECs seeded on the graphene–hemin device. In both (c,d), (A) 20 nM bradykinin, (B) 20 nM bradykinin and 1 mM L-NG-nitroarginine methyl ester (L-NAME), (C) without bradykinin.
As shown in Fig. 5a (A), after LPS (100 ng ml−1) was introduced (at t=1 h), the conductance of graphene–hemin device exhibited a gradual decrease, corresponding to an increase of NO concentration (Fig. 5b (A)). The NO concentration reached a maximum around t=6 h and remained stable for the following 5 h, followed by a slow decrease. This time course is consistent with the expected kinetics of NO generation by iNOS in macrophage cells46,47,48. In contrast, when LPS (100 ng ml−1) and aminoguanidine (an iNOS inhibitor; 100 μM) were added together, the change of conductance was almost negligible (Fig. 5a (B)). The conductance of the control device without LPS stimulation remained as the same value during the measurement (Fig. 5a (C)). As shown in Fig. 5b, the conductance of the device was converted to NO concentration by using the calibration curve of the graphene–hemin device (Supplementary Fig. S8, red curve). In general, the time response and overall concentration of NO are within the expected ranges for NO generated by macrophage cells. Moreover, the standard Griess test was also used to test NO released by the same batch of RAW 264.7 macrophage cells (Supplementary Fig. S9), which shows a consistent time course with that obtained from the electrical measurements. These results clearly demonstrate that the graphene–hemin sensors are capable of the real-time monitoring of extracellular NO generated from RAW 264.7 macrophage cells over a period of 20 h. Importantly, the calibration curve of the same device after the 20 h of continuous NO measurement shows that the sensitivity of the sensor is largely retained (Supplementary Fig. S8), further demonstrating the excellent long-term stability of the sensor.
We have further explored our sensor for the real-time detection of NO released from ECs. The production of NO by vascular ECs has an important role in normal vascular physiology50,51,52. Endothelial-derived NO can account for inhibition of platelet aggregation and adhesion and for the modulation of vascular tone. Direct measurement of NO released from ECs is difficult because NO is generated in small amounts and degrades fast in biological system. Importantly, we show that the graphene–hemin sensor can be readily used to measure NO released by human umbilical vein ECs (HUVECs). The HUVECs were seeded at 5 × 105 cells per ml (cell suspension) onto the graphene–hemin device on the SiO2 substrate (inset, Fig. 5d) within a PDMS well (set-up as in Supplementary Fig. S3). HUVECs were cultured for 12 h at 37 °C in a 5% CO2 incubator before the NO-sensing measurement.
The sensing measurement was carried out at 37 °C. With the introduction of bradykinin (20 nM, an agonist to stimulate the NO generation) at t=120 s, the conductance of graphene–hemin device started to decrease almost instantly (Fig.), and reached a plateau after about 150 s. The conductance then kept constant for about 600 s before a gradual increase to almost the original value in about another 200 s. As a control experiment, HUVECs were cultured with culture media with 1 mM L-NG-nitroarginine methyl ester, an endothelial NO synthase inhibitor, for 1 h before the electrical measurement. When bradykinin was added to the control sample, no apparent change of conductance was observed (Fig. 5c (B)). The conductance of another control device without bradykinin stimulation kept constant during the measurement (Fig. 5c (C)). Using the calibration curve of this graphene–hemin device (Supplementary Fig. S10), the conductance of the device can be converted to NO concentration (Fig. 5d). The concentration of NO started to increase after the injection of bradykinin and reached a plateau of ~20 nM in 150 s (Fig. 5d (A)). The time course and the concentration of NO are within the expected ranges of NO released by endothelial NO synthase50,51,52. These studies further demonstrate the capability of the graphene–hemin sensors to detect not only large amount of NO over a long period of time but also nanomolar range NO with fast response in complex biological systems.
Discussion
In brief, we have reported a new design of graphene–hemin FET-based NO sensors for direct electrical read-out of NO signals in real time. Unlike electrochemical sensors, the fundamental sensitivities of our graphene NO sensors are not directly dependent on their sizes. It can therefore allow us to scale the sizes of the sensors down to the lithographic limit while retaining high sensitivities. Importantly, our study demonstrates sub-nanomolar detection limit (0.3 nM) can be achieved with submicron graphene–hemin sensors (0.25 μm2), which can only be achieved with electrochemical sensors at least 4–5 orders of magnitude larger (1,500–30,000 μm2). Considering the two-dimensional nature of graphene materials, the miniaturized graphene NO sensor array can be readily integrated over large area for high-spatial resolution detection and imaging of NO signal. In addition, our graphene–hemin NO sensors can operate at a much lower bias voltage (for example, 10 mV) than the working potential of electrochemical NO sensors (for example, >0.6 V), and is therefore less invasive to biological systems.
We have shown that the tested NO sensors can exhibit high sensitivity in physiological solutions such as cell culture media or real biological samples such as fetal bovine serum, and can be used for the real-time monitoring of NO released by macrophage cells and ECs. All these samples represent complex real biological conditions with a large number of potentially interfering species at physiological levels. Altogether, these studies clearly demonstrate that our sensors can be used to detect NO in complex biological samples with excellent sensitivity, selectivity, as well as a rapid response speed. Along with their miniaturized sizes, these graphene–hemin NO sensors are promising as a new platform for highly sensitive real-time monitoring of NO signals in biological system with high spatiotemporal resolution.
Methods
Functionalization of graphene with hemin
The graphene devices were immersed in a 1-mg ml−1 hemin chloride/DMSO solution for 2 h to allow the non-covalent functionalization via π–π stacking. The sample was rinsed with DMSO for three times and isopropyl alcohol (IPA) for three times to wash away the unattached hemin molecules. After rinsing with IPA, the sample was blown dry with nitrogen gas.
Region selective functionalization
The graphene sample was first protected with a layer PMMA and a window was opened using e-beam lithography, and then immersed into hemin/methanol solution for 2 h. The sample was then taken out and rinsed with methanol and IPA. The sample was subsequently immersed in acetone and rinsed with IPA to remove the e-beam resist. Here methanol was used as the solvent to prevent the dissolution of e-beam resist PMMA by DMSO. The sensors functionalized in methanol solution respond to NO the same way as those functionalized in DMSO solution (Supplementary Fig. S11).
NO solution preparation
NO solution was prepared by dissolving an NO precursor (diethylamine NONOate sodium salt) into 10 mM NaOH solution followed by dilution with 10 mM PBS (pH=7.4), following a protocol developed in previous NO sensor studies15. All NaOH and PBS solutions were pre-bubbled with N2 for 2 h to remove the dissolved oxygen. Diethylamine NONOate sodium salt was added to a 10-mM NaOH solution to make a stock solution of 500 μM. Serial dilution was performed with the stock solution and 10 mM PBS (pH=7.4) to make NO solutions of various concentrations. The NO-containing PBS solutions were allowed to set for at least 15 min before the introduction into the PDMS channel to allow the NO concentrations to saturate. Griess test was used to confirm the concentration of NO generated by diethlyamine NONOate (Supplementary Fig. S12).
Device fabrication
Graphene was prepared by mechanical exfoliation onto a silicon substrate with a 300-nm thermal oxide layer. Titanium-gold thin film source, drain and solution gate electrodes were formed using e-beam lithography, vacuum metal deposition and a lift-off process. A typical graphene FET device has an active channel area ranging from 0.5 × 0.5 to 0.5 × 2 μm2, which is coupled with a PDMS microfluidic channel (with 0.5 mm channel height, 1 mm channel width and 10 mm channel length) for sample delivery.
Electrical measurement
The electrical transport measurements were conducted with a Lakeshore probe station (Model TTP4) and a computer-controlled analogue-to-digital converter (National Instruments model 6030E). All the sensing measurements were carried out under ambient conditions, with a pre-deposited Ti-Au pad as solution gate electrode.
Raw 264.7 cell culture
RAW 264.7 cells were obtained from the American Type Culture Collection; ATCC (Manassas, VA, USA) and cultured in DMEM; Carlsbad, CA, USA) containing 10% FCS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 2 mM L-glutamine (complete medium). These cells were maintained in 25 cm2 cell culture flasks, in which the cells were passaged at 70–80% confluency every 2–4 days.
HUVEC culture
HUVECs were obtained from the ATCC and cultured in F-12K Medium (ATCC) supplemented with 10% FCS, 0.1 mg ml−1 heparin; 0.05 mg ml−1 EC growth supplement. These cells were maintained in 25 cm2 cell culture flasks, in which the cells were passaged at 70–80% confluency every 5 days.
Additional information
How to cite this article: Jiang, S. et al. Real-time electrical detection of nitric oxide in biological systems with sub-nanomolar sensitivity. Nat. Commun. 4:2225 doi: 10.1038/ncomms3225 (2013).
References
Radomski, M. W. et al. An L-arginine nitric-oxide pathway present in human platelets regulates aggregation. Proc. Natl Acad. Sci. USA 87, 5193–5197 (1990).
Langrehr, J. M. et al. Nitric-oxide—a new endogenous immunomodulator. Transplantation 55, 1205–1212 (1993).
Ignarro, L. J. et al. Endothelium-derived relaxing factor produced and released from artery and vein is nitric-oxide. Proc. Natl Acad. Sci. USA 84, 9265–9269 (1987).
Ohta, A. et al. Localization of nitric-oxide synthase-immunoreactive neurons in the solitary nucleus and ventrolateral medulla-oblongata of the rat—their relation to catecholaminergic neurons. Neurosci. Lett. 158, 33–35 (1993).
Hall, C. N. & Garthwaite, J. What is the real physiological NO concentration in vivo? Nitric Oxide 21, 92–103 (2009).
Gryglewski, R. J., Palmer, R. M. J. & Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 320, 454–456 (1986).
Bedioui, F. & Villeneuve, N. Electrochemical nitric oxide sensors for biological samples—Principle, selected examples and applications. Electroanalysis 15, 5–18 (2003).
Kim, J. H. et al. The rational design of nitric oxide selectivity in single-walled carbon nanotube near-infrared fluorescence sensors for biological detection. Nat. Chem. 1, 473–481 (2009).
Kojima, H. et al. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal. Chem. 70, 2446–2453 (1998).
Wardman, P. et al. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: progress, pitfalls, and prospects. Free Radical Biol. Med. 43, 995–1022 (2007).
Bedioui, F. et al. Elaboration and use of nickel planar macrocyclic complex-based sensors for the direct electrochemical measurement of nitric oxide in biological media. Biosens. Bioelectron. 12, 205–212 (1997).
Lee, Y., Oh, B. K. & Meyerhoff, M. E. Improved planar amperometric nitric oxide sensor based on platinized platinum anode. 1. Experimental results and theory when applied for monitoring NO release from diazeniumdiolate-doped polymeric films. Anal. Chem. 76, 536–544 (2004).
Davies, I. R. & Zhang, X. J. Nitric oxide selective electrodes. Methods Enzymol. 436, 63–95 (2008).
Privett, B. J., Shin, J. H. & Schoenfisch, M. H. Electrochemical nitric oxide sensors for physiological measurements. Chem. Soc. Rev. 39, 1925–1935 (2010).
Wu, D. G. et al. Direct detection of low-concentration NO in physiological solutions by a new GaAs-based sensor. Chem. Eur. J. 7, 1743–1749 (2001).
Zheng, G. F. et al. Multiplexed electrical detection of cancer markers with nanowire sensor arrays. Nat. Biotechnol. 23, 1294–1301 (2005).
Chen, R. J. et al. An investigation of the mechanisms of electronic sensing of protein adsorption on carbon nanotube devices. J. Am. Chem. Soc. 126, 1563–1568 (2004).
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nat. Mater. 6, 183–191 (2007).
Castro Neto, A. H. et al. The electronic properties of graphene. Rev. Modern Phys. 81, 109–162 (2009).
Schwierz, F. Graphene transistors. Nat. Nanotech. 5, 487–496 (2010).
Liao, L. et al. High-kappa oxide nanoribbons as gate dielectrics for high mobility top-gated graphene transistors. Proc. Natl Acad. Sci. USA 107, 6711–6715 (2010).
Liao, L. & Duan, X. F. Graphene transistors for radio frequency electronics. Mater. Today 15, 328–337 (2012).
Weiss, N. O. et al. Graphene: an emerging electronic material. Adv. Mater. 24, 5782–5825 (2012).
Dresselhaus, M. S. & Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 51, 1–186 (2002).
Schedin, F. et al. Detection of individual gas molecules adsorbed on graphene. Nat. Mater. 6, 652–655 (2007).
Wehling, T. O. et al. Molecular doping of graphene. Nano Lett. 8, 173–177 (2008).
Dan, Y. P. et al. Intrinsic response of graphene vapor sensors. Nano Lett. 9, 1472–1475 (2009).
Mohanty, N. & Berry, V. Graphene-based single-bacterium resolution biodevice and DNA transistor: interfacing graphene derivatives with nanoscale and microscale biocomponents. Nano Lett. 8, 4469–4476 (2008).
Ang, P. K. et al. Solution-gated epitaxial graphene as pH sensor. J. Am. Chem. Soc. 130, 14392–14393 (2008).
Ohno, Y. et al. Electrolyte-gated graphene field-effect transistors for detecting pH protein adsorption. Nano Lett. 9, 3318–3322 (2009).
Cheng, Z. G. et al. Suspended graphene sensors with improved signal and reduced noise. Nano Lett. 10, 1864–1868 (2010).
Dong, X. C. et al. Electrical detection of DNA hybridization with single-base specificity using transistors based on CVD-grown graphene sheets. Adv. Mater. 22, 1649–1653 (2010).
Li, W. et al. Reduced graphene oxide electrically contacted graphene sensor for highly sensitive nitric oxide detection. ACS Nano 5, 6955–6961 (2011).
Xue, T. et al. Graphene-supported hemin as a highly active biomimetic oxidation catalyst. Angew. Chem. Intl. Ed. 51, 3822–3825 (2012).
Cooper, C. E. Nitric oxide and iron proteins. Biochim. Et Biophys. Acta Bioenerg. 1411, 290–309 (1999).
Xu, Y. et al. Chemically converted graphene induced molecular flattening of 5,10,15,20-tetrakis(1-methyl-4-pyridinio)porphyrin and its application for optical detection of cadmium(II) ions. J. Am. Chem. Soc. 131, 13490–13497 (2009).
Ferrari, A. C. Raman spectroscopy of graphene and graphite: disorder, electron-phonon coupling, doping and nonadiabatic effects. Solid State Commun. 143, 47–57 (2007).
Terner, J. et al. Resonance Raman spectroscopy of oxoiron(IV) porphyrin π-cation radical and oxoiron(IV) hemes in peroxidase intermediates. J. Inorg. Biochem. 100, 480–501 (2006).
Rao, C. N. R. & Voggu, R. Charge-transfer with graphene and nanotubes. Mater. Today 13, 34–40 (2010).
Maragos, C. M. et al. Complexes of NO with nucleophiles as agents for the controlled biological release of nitric-oxide—vasorelaxant effects. J. Med. Chem. 34, 3242–3247 (1991).
Hrabie, J. A. & John, R. K. New nitric oxide-releasing zwitterions derived from polyamines. J. Org. Chem. 58, 1472–1476 (1993).
Keefer, L. K. et al. “NONOates” ( 1-Substituted Diazen- 1-ium- 1,2- diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol. 268, 281–293 (1996).
Caraiman, D. et al. The gas-phase chemistry of iron cations coordinated to benzene and the extended aromatic coronene. Int. J. Mass Spectrom. 223, 411–425 (2003).
Hieringer, W. et al. The surface trans effect: influence of axial ligands on the surface chemical bonds of adsorbed metalloporphyrins. J. Amer. Chem. Soc. 133, 6206–6222 (2011).
Stuehr, D. J. & Marletta, M. A. Mammalian nitrate biosynthesis: mouse macrophages produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide. Proc. Natl Acad. Sci. USA 82, 7738–7742 (1985).
Cha, W. et al. Patterned electrode-based amperometric gas sensor for direct nitric oxide detection within microfluidic devices. Anal. Chem. 82, 3300–3305 (2010).
Pekarova, M. et al. Continuous electrochemical monitoring of nitric oxide production in murine macrophage cell line RAW 264.7. Anal. Bioanal. Chem. 394, 1497–1504 (2009).
Amatore, C. et al. Monitoring in real time with a microelectrode the release of reactive oxygen and nitrogen species by a single macrophage stimulated by its membrane mechanical depolarization. Chem. Bio. Chem 7, 653–661 (2006).
Palmer, R. M. J. et al. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 333, 664–666 (1988).
Malinski, T. & Taha, Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature 358, 676–678 (1992).
Pailleret, A. et al. In situ formation and scanning electrochemical microscopy assisted positioning of NO-sensors above human umbilical vein endothelial cells for the detection of nitric oxide release. Electrochem. Commun. 5, 847–852 (2003).
Acknowledgements
We thank the Nanoelectronics Research Facility (NRF) at UCLA for technical support. X.D. thanks the NIH Director’s New Innovator Award Program 1DP2OD004342-01 and partial support by Human Frontier Science Program. Y.H. thanks the NIH Director’s New Innovator Award Program 1DP2OD007279. A.N. thanks the National Science Foundation and the Environmental Protection Agency under Cooperative Agreement Number DBI 0830117 and the National Institute Of Environmental Health Sciences of the National Institutes of Health under Award Number U19ES019528.
Author information
Authors and Affiliations
Contributions
X.D. and Y.H. conceived and supervised the research. S.J. and R.C. performed most of the experiments (including device fabrication and measurements) and data analysis. X.W. conducted the cell culture experiments. T.X. contributed to surface functionalization. Y.L. contributed to device fabrication. X.D., Y.H. and A.N. supervised the research. X.D. and S.J. co-wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S12 and Supplementary Notes 1-3 (PDF 707 kb)
Rights and permissions
About this article
Cite this article
Jiang, S., Cheng, R., Wang, X. et al. Real-time electrical detection of nitric oxide in biological systems with sub-nanomolar sensitivity. Nat Commun 4, 2225 (2013). https://doi.org/10.1038/ncomms3225
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms3225
This article is cited by
-
Micro- and nano-devices for electrochemical sensing
Microchimica Acta (2022)
-
A DNA-based fluorescent probe maps NOS3 activity with subcellular spatial resolution
Nature Chemical Biology (2020)
-
Single-atom Ni-N4 provides a robust cellular NO sensor
Nature Communications (2020)
-
A Matrix Metalloproteinase Sensing Biosensor for the Evaluation of Chronic Wounds
BioChip Journal (2019)
-
Theoretical UV-Vis spectra of tetracationic porphyrin: effects of environment on electronic spectral properties
Journal of Molecular Modeling (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
