
Abstract
Graphene is a single layer of covalently bonded carbon atoms, which was discovered only 8 years ago and yet has already attracted intense research and commercial interest. Initial research focused on its remarkable electronic properties, such as the observation of massless Dirac fermions and the half-integer quantum Hall effect. Now graphene is finding application in touch-screen displays, as channels in high-frequency transistors and in graphene-based integrated circuits. The potential for using the unique properties of graphene in terahertz-frequency electronics is particularly exciting; however, initial experiments probing the terahertz-frequency response of graphene are only just emerging. Here we show that the photoconductivity of graphene at terahertz frequencies is dramatically altered by the adsorption of atmospheric gases, such as nitrogen and oxygen. Furthermore, we observe the signature of terahertz stimulated emission from gas-adsorbed graphene. Our findings highlight the importance of environmental conditions on the design and fabrication of high-speed, graphene-based devices.
Similar content being viewed by others
Introduction
The experimental realization of graphene, a single layer of graphite, in 2004 (ref.1) rapidly led to the observation of massless Dirac fermions and the half-integer quantum Hall effect2. Since then, many potential applications have been suggested3,4, and recently graphene-based touch-screen displays5, transistors6,7 and integrated circuits8 have been demonstrated. The high mobility of charge carriers in graphene makes it an ideal material for extremely high-speed devices6,7, indeed terahertz-frequency switching has been predicted3. The potential for the realization of such devices was greatly enhanced by the emergence of large-area graphene sheets grown by chemical vapour deposition (CVD)9. However, the design and production of these high-frequency devices require a fuller understanding of both non-equilibrium carrier dynamics in graphene10 and its susceptibility to environmental conditions11.
Ultrafast spectroscopic techniques provide an excellent probe of non-equilibrium carrier dynamics in graphene12. Ultrafast cooling and relaxation of photoexcited charge carriers in graphene have been observed using optical pump–optical probe spectroscopy. Several studies10,13,14 have shown that graphene becomes more transparent at optical frequencies after intense photoexcitation (photo-induced bleaching), owing to state blocking of interband absorption. Furthermore, these studies showed that thermalization of photoexcited carriers occurs within tens of femtoseconds, and a subsequent cooling by carrier–phonon interactions occurs on a picosecond time scale.
Although optical photons probe interband charge dynamics, terahertz frequency photons are excellent probes of intraband dynamics. Studies of intraband relaxation in graphene have been performed using optical pump–terahertz probe spectroscopy12,15,16,17. George et al.15 revealed that the absorption of the terahertz photons in epitaxially grown graphene increases after intense (interband) optical excitation. The increase in absorption was attributed to an increase in the density of mobile charge carriers after photoexcitation. However, a comparable study on CVD-grown graphene found the opposite effect photo-induced bleaching18. It is known that CVD-grown graphene is p-doped, due to residual impurities and defects from the production process19, unlike epitaxial graphene17. Despite this, equilibrium carrier dynamics in CVD-grown graphene have been shown to be similar to other forms of graphene in the terahertz regime, suggesting that growth conditions cannot explain the discrepancy between the epitaxial and CVD graphene in the pump–probe studies20.
In this study, we utilized terahertz time-domain spectroscopy to measure the photoconductivty of CVD-grown graphene in four different environment types: air, oxygen, nitrogen and vacuum. We demonstrate that CVD-grown graphene can display either photo-induced bleaching or photo-induced absorption, depending on the type and density of gas adsorbed on its surface. Our results highlight the dramatic effects environmental conditions have on the high-frequency electrical properties of graphene. These results have important implications for the design of future graphene electronic devices. In particular, the order-of-magnitude changes in photoconductivity we observe could provide a basis for precise graphene-based gas monitors. Conversely, the sensitivity of the opto-electronic properties of graphene to environmental conditions will need to be addressed during the design and fabrication of future devices.
Results
Sample preparation and characterization
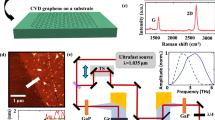
The CVD samples used in this study were grown on copper foil and transferred to z-cut quartz substrates using PMMA (poly(methyl methacrylate)) as a supporting layer (for details see the Methods section). Raman microscopy and atomic force microscopy (AFM) revealed that the samples were primarily a graphene monolayer with relatively few defects. A typical Raman spectrum is shown in Fig. 1 and exhibits a Lorentzian 2D Raman peak (2,700 cm−1), with the ratio of intensities of the 2D and G (1,580 cm−1) peaks, I2D/IG~2, which is indicative of monolayer growth. In addition, the relative weakness of the D peak (1,350 cm−1) demonstrates the small number of defects and low disorder in the samples, confirming their high quality21. Ultraviolet-visible absorption measurements revealed an absorption of ~πα, where α is the fine structure constant, also suggesting monolayer growth (see Supplementary Methods)22. The AFM images (Fig. 1b) showed the graphene sheet thickness measured from the edge to be ~0.72 nm, further suggesting monolayer growth23. Hall measurements revealed the sample to be p-doped, with a hole density 8 × 1012 cm−2 and Fermi level εF ~−0.3 eV.

(a) Raman spectroscopy. The relative intensities of the 2D and G peaks suggest monolayer graphene21. Inset: photograph of graphene sheet with an area of 1 × 1 cm2 on quartz. (b) Surface topography of CVD-grown graphene measured by AFM. The residuals remaining on graphene surface can be attributed to incomplete decomposition or carbonization of PMMA.
Terahertz time-domain spectroscopy
To assess the influence of atmospheric gas adsorption on graphene, we used terahertz spectroscopy. This technique allowed us to observe intraband charge dynamics and investigate the high-frequency electrical properties of graphene. Terahertz spectroscopy is ideal for electrically characterizing surface-sensitive materials such as graphene, as it is a non-contact technique and covers an important frequency range (~100 GHz to >3 THz) for high-speed device operation24. Optical pump–terahertz probe spectroscopy measurements were performed with CVD-grown graphene samples in an atmosphere of air, N2 or O2, or in a vacuum. Before each set of measurements, the sample was left under a vacuum of 10−5 mbar for 2 h to remove existing adsorbants (see Supplementary Methods for Langmuir coverage calculations). For the vacuum measurements, the graphene was left for a further 2 h with 800 nm (1.55 eV) ‘pump’ photons incident upon the sample before measurement. For the gaseous measurements, 1 bar of air, N2 or O2, was introduced after 2 h under vacuum, and measurements were taken after a further 2 h, again with the ‘pump’ beam incident upon the sample. Measurements were found to be reproducible on different samples and were independent of sample history. That is, the effect of exposure to any of the gases could be ‘reset’ by placing the sample in vacuum for 2 h.
For the measurements, the electric field of a terahertz pulse transmitted through the graphene was sampled after photoexcitation by an 800 nm, 180 μJ cm−2 fluence pump pulse,  , and without the photoexcitation,
, and without the photoexcitation,  . The photo-induced change in terahertz transmission of the sample,
. The photo-induced change in terahertz transmission of the sample,  was recorded using lock-in detection. The principle of the measurements is illustrated in Fig. 2a. The terahertz pulse probes the photoexcited graphene sample at a time t2 after photoexcitation. The terahertz probe is then mapped out in time by varying the difference in time of arrival, t1, between the terahertz pulse and an 800-nm gate pulse at an electro-optic detection crystal (GaP).
was recorded using lock-in detection. The principle of the measurements is illustrated in Fig. 2a. The terahertz pulse probes the photoexcited graphene sample at a time t2 after photoexcitation. The terahertz probe is then mapped out in time by varying the difference in time of arrival, t1, between the terahertz pulse and an 800-nm gate pulse at an electro-optic detection crystal (GaP).

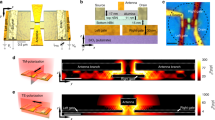
(a) Schematic representation of the measurement. Varying t1 allows the electric field, ETHz, of the terahertz probe pulse to be sampled in the electro-optic (E-O) crystal. The sample can be probed at different times after photoexcitation by altering t2. (b,c) Terahertz electric field, ETHz, transmitted through the sample (b), and the corresponding photo-induced change in transmission, ΔE (c), as a function of the pump–probe delay time, t2. (d,e) Real (d) and imaginary (e) parts of the frequency-dependent photoconductivity as a function of t2. The contours highlight increments of 2 × 10−5 (Ω/ )−1. The thicker contour in e represents Im(Δσ)=0, corresponding to the Lorentzian peak frequency, ω0. Measurements were performed in pure oxygen at room temperature and pressure.
)−1. The thicker contour in e represents Im(Δσ)=0, corresponding to the Lorentzian peak frequency, ω0. Measurements were performed in pure oxygen at room temperature and pressure.
In Fig. 2b we present the measured terahertz electric field, ETHz, transmitted through graphene in a pure oxygen atmosphere and the corresponding change in transmission due to photoexcitation, ΔE. The induced ΔE decreases as the delay between pump and probe, t2, is increased. The photo-induced change in terahertz transmission is seen to decay within ~5 ps. Further information may be extracted from these data by performing a Fourier transform on the electric field data to give directly the frequency-dependent complex photoconductivity, Δσ(ω), without resorting to the Kramers–Kronig relations, via17

where Z0 is the impedance of free space, Z0=377 Ω and ns is the refractive index of the quartz substrate, nr=2.1 (25) at terahertz frequencies. Figure 2d show the real and imaginary parts of the photoconductivity, respectively, as a function of the pump–probe delay time.
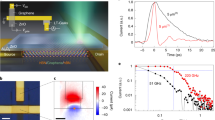
To more easily compare the photoconductivity in different atmospheres, we take slices of the data from Fig. 2 and the corresponding data for the other environment types. For instance, holding t1 constant at the maximum terahertz electric field (for example, t1=0 in Fig. 2c) and observing the resulting ΔE as pump–probe delay time t2 is varied yields an optical pump–terahertz probe measurement, which provides the average terahertz response of the sample. In Fig. 3, we plot −ΔE/ETHz, which is proportional to the photoconductivity of the graphene, as a function of the delay time between the pump and probe pulses in the four atmosphere types. It should be noted that because of the large number of holes already present near the Dirac point in this p-doped sample, the transient response probed by the terahertz is dominated by electron dynamics.

Here,  . From top to bottom: graphene in vacuum, N2, air and O2 at room temperature. Photo-induced absorption is observed in vacuum, but photo-induced bleaching is observed in gaseous environments. Inset: schematic representation of the pump–probe measurements of p-doped graphene (εF~0.3 eV) in vacuum (top) and in the presence of gases (bottom), with corresponding density of states (g(ε)). The high-energy pump (
. From top to bottom: graphene in vacuum, N2, air and O2 at room temperature. Photo-induced absorption is observed in vacuum, but photo-induced bleaching is observed in gaseous environments. Inset: schematic representation of the pump–probe measurements of p-doped graphene (εF~0.3 eV) in vacuum (top) and in the presence of gases (bottom), with corresponding density of states (g(ε)). The high-energy pump ( ) excites carriers far above the Dirac point, which cool by phonon emission. In gapless graphene, terahertz photons can be absorbed by the photoexcited carriers. In the presence of gases, stimulated emission releases extra terahertz photons, yielding a bleaching pump–probe signal.
) excites carriers far above the Dirac point, which cool by phonon emission. In gapless graphene, terahertz photons can be absorbed by the photoexcited carriers. In the presence of gases, stimulated emission releases extra terahertz photons, yielding a bleaching pump–probe signal.
Immediately after photoexcitation in vacuum, the terahertz conductivity increases (photo-induced absorption) within 1 ps, and subsequently relaxes with two distinct time scales. Initially, fast cooling occurs within ~0.6 ps and a longer, exponential cooling occurs with a time scale of ~2.5 ps. Such a relaxation is consistent with other studies, which associated the first rapid cooling with emission of optical phonons, followed by a bottleneck when the carrier temperature cools below the hot optical phonon temperature (~0.196 and 0.162 eV (ref. 16)), slowing further cooling10,14,16.
However, when graphene is placed in a gaseous atmosphere, the photo-induced change is dramatically different. Surprisingly, the photoconductivity signal flips from being purely positive in vacuum to purely negative in all the gas environments. Following photoexcitation in air, N2 or O2, the terahertz conductivity decreases within 1 ps, and then relaxes with a common monoexponential time scale of 2.5 ps. The similarity between this time constant and that of cooling in vacuum suggests the continued importance of the carrier–phonon interactions. Adsorbed O2 was found to have the strongest influence on charge dynamics in graphene, with the negative photoconductivity almost an order of magnitude greater than the induced absorption under vacuum. A negative transient absorption, such as we observe, may be attributed to either stimulated emission in the samples or a bleaching effect. To understand more about the origin of the negative photoconductivity, it is informative to consider the photoconductivity spectra at a fixed time after photoexcitation.
Figure 4 shows the photoconductivity in all four atmosphere types at 2 ps after photoexcitation (t2=2 ps). The spectrum of graphene in vacuum reveals a relatively flat and positive real part of photoconductivity, in stark contrast to the strongly negative (real) photoconductivity seen for gas-absorbed graphene. Furthermore, the spectral photoconductivity of graphene samples in the presence of N2, air and O2 can be seen to exhibit a Lorentzian form,

(a–d) Photoconductivity in vacuum (a), N2 (b), air (c) and O2 (d), taken at 2 ps after photoexcitation by the optical pump. Solid orange dots show the real part of the photoconductivity, hollow blue dots show the imaginary part. The solid black line in b–d is a Lorentzian model fit to the conductivity data, demonstrating the Lorentzian form the of photoconductivity in gaseous environments. Shading is used to emphasize the change from positive to negative Im(Δσ). This point corresponds to the peak Lorentzian frequency, ω0.

where Γ is the linewidth and ω0 is the resonant frequency. The solid lines in Fig. 4 represent a least square fit of equation 2 to the experimental data. The fitted resonant frequency for graphene in air and N2 was found to be ω0~1.8 THz compared with ω0~2 THz for graphene in an O2 environment. The resonance in all gas types is spectrally very broad; the fitted linewidth was Γ~10 THz, independent of the gas type. The Lorentzian fit begins to deviate from the measured photoconductivity at low frequencies. The deviation is most noticeable for O2-exposed graphene (Fig. 4d). The non-zero value of Re(Δσ) evident in the data at low frequency suggests an underlying Drude-like response; however, inclusion of a Drude term does not enhance the fit within our observable bandwidth, and so has been neglected. In contrast, the photo-induced absorption in vacuum leads to a small, positive, spectrally flat, real conductivity, with a non-negligible imaginary part.
A negative transient absorption signal with a Lorentzian lineshape is characteristic of stimulated emission. Hence, our data provide evidence that stimulated emission of terahertz photons is occurring in photoexcited graphene in the presence of atmospheric gases. Theoretical studies have predicted amplified stimulated emission of terahertz radiation from sufficiently intensely photoexcited graphene26. The proposed mechanism is that after photoexcitation, the charge carriers relax and populate states around the Dirac point from where they can radiatively recombine. Thus, terahertz emission over a large spectral range is expected to be possible26. Although our measurements do not show amplification of the terahertz probe (losses are still greater than gain), stimulated terahertz emission below the gain threshold is consistent with our observations.
Discussion
We now consider the mechanism for the dramatic changes of photoconductivity in graphene under different gas environments. The ability to ‘reset’ the effects of gas exposure by placing the graphene samples in vacuum for 2 h (at room temperature) suggests that the gas molecules are physisorbed rather than chemisorbed to the graphene surface. Physisorption of O2 and N2 on graphene could act as a dopant27, lead to the opening of a small band gap at the Dirac point28,29 and/or lead to a modification of the density of states30,31. Hall measurements (see Methods) showed only 3% changes in the Fermi level after gas adsorption, indicating that doping alone is unlikely to account for the huge change in photoconductivity we observe. The opening of an ~8-meV band gap on gas adsorption and associated increase in the density of electron and hole states near the band extrema would be consistent with our observations. In this case, electrons injected into the conduction band by 800 nm (1.55 eV) photoexcitation would relax rapidly to near the band minimum via phonon-mediated processes, as shown on the inset of Fig. 3. Recombination for the photoinjected electrons across the band gap with a sea of holes would lead to the observed stimulated emission signal. The spectral broadness (Γ~10 THz) of the observed transition may then be explained by a combination of the following factors: a high interband transition rate (lifetime broadening), fast dephasing via electron–electron interactions and a cooling bottleneck below the optical phonon energies. However, full ab-inito calculations would be required to confirm the exact mechanism. Note that as the CVD graphene is heavily p-doped, the opening of a band gap is not directly observable by terahertz transmission spectroscopy, as there are no electrons near the band gap to be excited by the low-energy terahertz photons (see Supplementary Discussion and inset of Fig. 3).
The adsorption of molecules on graphene would locally open a band gap and hence alter the otherwise linear density of states (g(ε)) of the graphene, thereby increasing g(ε) at the top of the valance band and bottom of the conduction band (see inset of Fig. 3)31. However, a gap would only be opened in regions around adsorbed gas molecules, creating islands of semiconducting graphene amongst the unaltered, gapless graphene (see Supplementary Methods for Langmuir coverage calculations). As the terahertz probe is much larger than these puddles, the observed photoconductivity behaviour is the result of an interaction with both types of graphene. Increased stimulated emission will therefore be observed if gas molecules are more effectively adsorbed on the surface, thus creating a higher proportion of semiconducting material. Of the gases used in this study, O2 is most likely to interact with graphene32,33, which is consistent with the increased negative photoconductivity signal observed in pure O2 compared with air and N2.
There are a number of other possible explanations for a Lorentzian photoconductivity in graphene. A positive Lorentzian conductivity may originate from the excitation of plasmons34. Terahertz surface plasmons have been predicted theoretically35,36 and recently observed in spatially confined graphene37. However, the peak frequency of a plasmon-dependent conductivity varies with carrier concentration as ω0 n1/4 (ref. 37), and Re(Δσ) is expected to be positive in contrast to our experiments. Figure 2 demonstrates that our measured ω0 does not decrease with time after photoexcitation, and hence is independent of photoexcited carrier concentration. Additional measurements showed ω0 to be independent of pump fluence (see Supplementary Discussion). Thus, it is unlikely that excitation of plasmons explains the Lorentzian response. A negative photoconductivity may also be attributed to a photo-induced reduction in conductivity, for example, by an increase in the carrier–carrier or carrier–phonon (intraband) scattering rates. However, it is not clear why such mechanisms would produce a Lorentzian form or occur only in gas-adsorbed graphene. Finally, ground-state bleaching could lead to a negative photoconductivity with Lorentzian form; however, the states associated with the absorption of (1.55 eV) optical photons in graphene are well separated from the states near the Dirac point that are associated with interband terahertz absorption and emission (see the inset of Fig. 3). We therefore conclude that the opening of a small band gap in the presence of adsorbed gas molecules is the most likely explanation for our measurements.
n1/4 (ref. 37), and Re(Δσ) is expected to be positive in contrast to our experiments. Figure 2 demonstrates that our measured ω0 does not decrease with time after photoexcitation, and hence is independent of photoexcited carrier concentration. Additional measurements showed ω0 to be independent of pump fluence (see Supplementary Discussion). Thus, it is unlikely that excitation of plasmons explains the Lorentzian response. A negative photoconductivity may also be attributed to a photo-induced reduction in conductivity, for example, by an increase in the carrier–carrier or carrier–phonon (intraband) scattering rates. However, it is not clear why such mechanisms would produce a Lorentzian form or occur only in gas-adsorbed graphene. Finally, ground-state bleaching could lead to a negative photoconductivity with Lorentzian form; however, the states associated with the absorption of (1.55 eV) optical photons in graphene are well separated from the states near the Dirac point that are associated with interband terahertz absorption and emission (see the inset of Fig. 3). We therefore conclude that the opening of a small band gap in the presence of adsorbed gas molecules is the most likely explanation for our measurements.
In summary, we have used terahertz time-domain spectroscopy to non-invasively probe photoexcited carrier dynamics in CVD-grown graphene. We have observed that gas adsorption can dramatically alter the high-frequency electrical response of graphene sheets. We also provide experimental evidence for stimulated emission of terahertz photons in graphene. Furthermore, our results demonstrate the huge influence environmental factors can exert on the behaviour of hot carriers in CVD graphene, and highlight the importance of consideration of such factors for future CVD graphene opto-electronic devices.
Methods
Synthesis and transfer of CVD graphene
Large-area graphene films were prepared by CVD on 25 μm copper foil (Alfa Aesar). Before the growth, the copper foil was heated in hydrogen from room temperature to 1,000 °C for 60 min for removal of surface oxides. As temperature reached 1,000 °C, a mixed flow of H2 (15 sccm) and CH4 (60 sccm) was introduced for graphene growth. After CVD, the graphene film was separated from copper foil and then attached to the z-cut quartz substrate by the polymer transfer process: PMMA; MicroChem Co., NANO PMMA 950K A4) was spin-coated on graphene/copper foil to form a protective layer. The copper foil was etched at 25 °C in ferric nitride solution (50 g l−1, J.T. Baker ACS reagent, 98%), leaving a transparent PMMA/graphene film floating on the solution. After rinsing, the film was transferred onto the quartz substrate and the PMMA capping layer was dissolved in acetone at 60 °C overnight. To remove the residual PMMA, graphene films were annealed at 450 °C at 500 Torr in the mixed gases of H2 (20 sccm) and Ar (80 sccm) for decomposition of the polymer.
Characterization
Primary characterization of the sample was performed by Raman spectroscopy21, using an NT-MDT confocal Raman microscopic system (laser wavelength 473 nm, power 0.5–1 mW, spot size 0.5 μm).
Additional characterization was performed using ultraviolet-visible absorption spectroscopy (Perkin-Elmer Lambda 9 UV-visible-NIR Spectrophotometer) in the range 500–1,300 nm, confirming the expected constant πα absorption of a single layer of graphene in this wavelength range22.
Nominally identical samples were characterized using AFM (Veeco Dimension-Icon) and room temperature Hall effect measurements based on the van der Pauw method (3S Co., NI PXI-1033). From the Hall effect measurements averaged across eight samples, the sheet resistance, carrier density and Hall mobility were found to be 1,100±350 Ω/□, 8±2 × 1012 cm−2 and 800±250 cm2 V s−1, respectively. The characterizations show that the quality of the graphene samples is fairly good.
Further Hall effect measurements were performed to examine the equilibrium electrical properties of the graphene samples under different gaseous environments. Two systems were used for the measurements, one based on a permanent magnet (0.68 T), and another using a swept electromagnet (0–0.91 T). Samples were held in a vacuum vessel, which could be purged with different gasses. The Fermi levels were deduced using the relation  , where vF≈106 ms−1(ref. 2,37).
, where vF≈106 ms−1(ref. 2,37).
The equilibrium carrier density, and hence Fermi level, were found to be relatively insensitive to the gaseous environment, with variations in n and εF between samples being greater than changes induced by the gases. It was found that compared with vacuum conditions, exposure to pure nitrogen gas decreased the carrier density by ~5%, exposure to oxygen increased the carrier density by ~7% and exposure to air increased it by ~3%, correspnding to Fermi level changes of ~2%, ~3% and ~1%, respectively. These changes are consistent with previous observation of oxygen and nitrogen exposure doping CVD graphene p-type and n-type, respectively27,38.
Terahertz spectroscopy
A Ti:sapphire regenerative amplifier (Spectra-Physics Spitfire Pro) was used as the pulse laser source for both terahertz generation and optical pumping of the sample: 800 nm, 45 fs pulse duration, 5 kHz repetition rate and 4 W average power. Of this beam, 2.9 W, reduced to 0.29 W with a neutral density filter, was used to photoexcite the graphene sample, with a maximum fluence of 180 μJ cm−2 incident upon the sample, and beam width 2 mm.
A fraction (200 μJ per pulse) of the amplifier beam was used to generate the terahertz probe beam by optical rectification24 in a 2-mm GaP crystal. The terahertz beam had a width of 0.48 mm at the sample position. The area of graphene probed by the terahertz beam was much smaller than the area photoexcited by the pump beam; thus, the probe measured an area of roughly constant photoexcited carrier density.
The remainder of the amplifier beam (16 μJ per pulse) was used as a gate beam for electro-optic sampling24 of the terahertz field in a 200-μm GaP crystal. The terahertz electric field, ETHz, was detected by a balanced photodiode circuit, and the signal extracted using a lock-in amplifier (Stanford Research SR830), referenced to a 2.5-kHz chopper in the terahertz generation beam. A further lock-in (SR830) was used to detect the optical pump-induced change in ETHz, ΔE, by referencing to a 125-Hz chopper in the sample pump beam.
The gaseous atmospheres used in the terahertz measurements were ambient air, 99.998% pure oxygen-free N2 and 99.99% pure O2. A vacuum of 10−5 mbar, generated using an oil-free pumping system (turbo molecular pump and scroll pump), was used for both the vacuum measurements and to remove adsorbants from the sample before gaseous measurements.
Additional information
How to cite this article: Docherty, C. J. et al. Extreme sensitivity of graphene photoconductivity to environmental gases. Nat. Commun. 3:1228 doi: 10.1038/ncomms2235 (2012).
References
Novoselov K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Novoselov K. S. et al. Two-dimensional gas of massless dirac fermions in graphene. Nature 438, 197–200 (2005).
Geim A. K. Graphene: status and prospects. Science 324, 1530–1534 (2009).
Bao Q., Loh K. P. Graphene photonics, plasmonics, and broad optoelectronic devices. ACS Nano 6, 3677–3694 (2012).
Bae S. et al. Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat. Nanotechnol. 5, 574–578 (2010).
Lin Y. M., Jenkins K. A., Valdes-garcia A., Small J. P., Farmer D. B., Avouris P. Operation of graphene transistors at gigahertz frequencies. Nano Lett. 9, 422–426 (2009).
Wu Y. Q. et al. High-frequency, scaled graphene transistors on diamond-like carbon. Nature 472, 74–78 (2011).
Lin Y. M. et al. Wafer-scale graphene integrated circuit. Science 332, 1294–1297 (2011).
Li X. S. et al. Large-area synthesis of high-quality and uniform graphene films on copper foils. Science 324, 1312–1314 (2009).
Wang H. N. et al. Ultrafast relaxation dynamics of hot optical phonons in graphene. Appl. Phys. Lett. 96, 081917 (2010).
Klarskov M. B. et al. Fast and direct measurements of the electrical properties of graphene using micro four-point probes. Nanotechnology 22, 445702 (2011).
Docherty C. J., Johnston M. B. Terahertz properties of graphene. J. Infrared Millim. Terahertz Waves 33, 797–815 (2012).
Dawlaty J. M., Shivaraman S., Chandrashekhar M., Rana F., Spencer M. G. Measurement of ultrafast carrier dynamics in epitaxial graphene. Appl. Phys. Lett. 92, 042116 (2008).
Hale P. J., Hornett S. M., Moger J., Horsell D. W., Hendry E. Hot phonon decay in supported and suspended exfoliated graphene. Phys. Rev. B 83, 121404 (2011).
George P. A. et al. Ultrafast optical-pump terahertz-probe spectroscopy of the carrier relaxation and recombination dynamics in epitaxial graphene. Nano Lett. 8, 4248–4251 (2008).
Strait J. H., Wang H. N., Shivaraman S., Shields V., Spencer M., Rana F. Very slow cooling dynamics of photoexcited carriers in graphene observed by optical-pump terahertz-probe spectroscopy. Nano Lett. 11, 4902–4906 (2011).
Choi H. et al. Broadband electromagnetic response and ultrafast dynamics of few-layer epitaxial graphene. Appl. Phys. Lett. 94, 172102 (2009).
Hwang H. Y., Brandt N. C., Hootan F., Hsu A. L., Kong J., Nelson. K. A. Nonlinear THz conductivity dynamics in CVD-grown graphene Preprint at http://arxiv.org/abs/1101.4985 (2011).
Horng J. et al. Drude conductivity of dirac fermions in graphene. Phys. Rev. B 83, 165113 (2011).
Lee C., Kim J. Y., Bae S., Kim K. S., Hong B. H., Choi E. J. Optical response of large scale single layer graphene. Appl. Phys. Lett. 98, 071905 (2011).
Ferrari A. C. et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 97, 187401 (2006).
Nair R. R. et al. Fine structure constant defines visual transparency of graphene. Science 320, 1308 (2008).
Gupta A., Chen G., Joshi P., Tadigadapa S., Eklund. P. C. Raman scattering from high-frequency phonons in supported n-graphene layer films. Nano Lett. 6, 2667–2673 (2006).
Ulbricht R., Hendry E., Shan J., Heinz T. F., Bonn M. Carrier dynamics in semiconductors studied with time-resolved terahertz spectroscopy. Rev. Mod. Phys. 83, 543–586 (2011).
Grischkowsky D., Keiding S., van Exter M., Fattinger C. Far-infrared time-domain spectroscopy with terahertz beams of dielectrics and semiconductors. J. Opt. Soc. Am. B Opt. Phys. 7, 2006–2015 (1990).
Ryzhii V., Ryzhii M., Otsuji T. Negative dynamic conductivity of graphene with optical pumping. J. Appl. Phys. 101, 083114 (2007).
Shi Y. M., Fang W. J., Zhang K. K., Zhang W. J., Li L. J. Photoelectrical response in single-layer graphene transistors. Small 5, 2005–2011 (2009).
Ito J., Nakamura J., Natori A. Semiconducting nature of the oxygen-adsorbed graphene sheet. J. Appl. Phys. 103, 113712 (2008).
Berashevich J., Chakraborty. T. Tunable band gap and magnetic ordering by adsorption of molecules on graphene. Phys. Rev. B 80, 033404 (2009).
Dai J. Y., Yuan J. M. Adsorption of molecular oxygen on doped graphene: atomic, electronic, and magnetic properties. Phys. Rev. B 81, 165414 (2010).
Ando T. The electronic properties of graphene and carbon nanotubes. NPG Asia Mater. 1, 17–21 (2009).
Ryu S. et al. Atmospheric oxygen binding and hole doping in deformed graphene on a SiO2 substrate. Nano Lett. 10, 4944–4951 (2010).
Sato Y., Takai K., Enoki T. Electrically controlled adsorption of oxygen in bilayer graphene devices. Nano Lett. 11, 3468–3475 (2011).
Parkinson P. et al. Carrier lifetime and mobility enhancement in nearly defect-free core-shell nanowires measured using time-resolved terahertz spectroscopy. Nano Lett. 9, 3349–3353 (2009).
Rana F. Graphene terahertz plasmon oscillators. IEEE Trans. Nanotechnol. 7, 91–99 (2008).
Ryzhii V., Satou A., Otsuji T. Plasma waves in two-dimensional electron-hole system in gated graphene heterostructures. J. Appl. Phys. 101, 024509 (2007).
Ju L. et al. Graphene plasmonics for tunable terahertz metamaterials. Nat. Nanotechnol. 6, 630–634 (2011).
Daas B. K., Nomani W. K., Daniels K. M., Sudarshan T. S., Koley G., Chandrashekhar M. V. S. Molecular gas adsorption induced carrier transport studies of epitaxial graphene using IR reflection spectroscopy Preprint at http://arxiv.org/abs/1201.4746 (2012).
Acknowledgements
We thank the EPSRC (UK) for financial support.
Author information
Authors and Affiliations
Contributions
C.J.D. performed the terahertz measurements and data analysis. C.T.L. and L.J.L. grew and processed the graphene samples and performed Raman spectroscopy. C.J.D., H.J.J. and R.J.N. performed Hall measurements in Oxford. C.T.L. and L.J.L. performed Hall measurements in Taipei. M.B.J. conceived the study and all authors discussed the results. C.J.D. and M.B.J. wrote the manuscript, supported by L.M.H. and R.J.N.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S6, Supplementary Table S1, Supplementary Discussion, Supplementary Methods and Supplementary References (PDF 1493 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Docherty, C., Lin, CT., Joyce, H. et al. Extreme sensitivity of graphene photoconductivity to environmental gases. Nat Commun 3, 1228 (2012). https://doi.org/10.1038/ncomms2235
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms2235
This article is cited by
-
Intense terahertz radiation: generation and application
Frontiers of Optoelectronics (2021)
-
Significant enhancement in the hydrogen-sensing performance of polypyrrole/titanium oxide (PPy/TiO2) hybrid sensors by a chemical oxidation polymerization approach
Journal of Materials Science: Materials in Electronics (2020)
-
Colorless-to-colorful switching electrochromic polyimides with very high contrast ratio
Nature Communications (2019)
-
Decrease in Terahertz Conductivity of Graphene Under Electron Beam Irradiations
Journal of Infrared, Millimeter, and Terahertz Waves (2019)
-
In-situ terahertz optical Hall effect measurements of ambient effects on free charge carrier properties of epitaxial graphene
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
