
Abstract
Topological insulators are characterized by a non-trivial band topology driven by the spin-orbit coupling. To fully explore the fundamental science and application of topological insulators, material realization is indispensable. Here we predict, based on tight-binding modelling and first-principles calculations, that bilayers of perovskite-type transition-metal oxides grown along the [111] crystallographic axis are potential candidates for two-dimensional topological insulators. The topological band structure of these materials can be fine-tuned by changing dopant ions, substrates and external gate voltages. We predict that LaAuO3 bilayers have a topologically non-trivial energy gap of about 0.15 eV, which is sufficiently large to realize the quantum spin Hall effect at room temperature. Intriguing phenomena, such as fractional quantum Hall effect, associated with the nearly flat topologically non-trivial bands found in eg systems are also discussed.
Similar content being viewed by others


Introduction
Since the discovery1,2 of the quantum Hall effect (QHE), the quest for topologically ordered states of matter has become a major subject of interest in condensed matter physics. Haldane3 first proposed that electrons hopping on a honeycomb lattice could realize the QHE in the absence of Landau levels, pointing out the possibility of non-trivial topology in simple band insulators. Along this direction, recent efforts have culminated in the theoretical prediction4,5,6,7 and subsequent experimental realization8,9,10 of the so-called topological insulators (TIs) in materials with strong spin-orbit coupling (SOC). Many interesting phenomena, including giant magneto-electric effects11 and the appearance of Majorana fermions12, have been predicted. Once realized in real materials, these phenomena could lead to entirely new device paradigms for spintronics and quantum computing.
However, so far, the material realization of TIs has been limited to narrow band-gap semiconductors based on Hg or Bi, in which the electronic properties are dominated by s and p orbitals. Here, we report our theoretical investigation of topological insulating behaviour in a completely different materials class—heterostructures of transition-metal oxides (TMOs) involving d electrons. Our motivation is twofold. First, artificial heterostructures of TMOs are becoming available owing to the recent development13,14,15 in the fields of oxide superlattices and oxide electronics16. In particular, layered structures of TMOs can be now prepared with atomic precision, thus offering a high degree of control over important material properties, such as lattice constant, carrier concentration, SOC and correlation strength. As we show below, these advantages can be readily exploited in the design of TIs. Second, TMOs constitute a wide class of compounds that exhibit a variety of intriguing properties and electronic states associated with the electron–electron interactions, encompassing superconductivity, magnetism, ferroelectricity and Mott insulators. Combined with the TI phase, TMO heterostructures provide a very promising platform to explore various topological effects.
Our main results are summarized below. We first demonstrate the design principle for realizing two-dimensional (2D) TIs in bilayers of perovskite-type TMOs grown along the [111] crystallographic axis by using phenomenological tight-binding (TB) modelling. Based on this design principle and first-principles calculations, a number of candidate materials are identified. The topological band structure of these materials can be fine-tuned by changing dopant ions, substrates and external gate voltages, which will enable also the control of the topological quantum phase transition. In particular, we predict that LaAuO3 bilayer has a topologically non-trivial energy gap about 0.15 eV, which is sufficiently large to realize the quantum spin Hall effect at room temperature. When electron–electron interaction is included, our system with topologically non-trivial band structure could have far more interesting physics. Here, we demonstrate this possibility by focusing on the TMO bilayers of eg systems, which are characterized by nearly flat topologically non-trivial Z2 bands. We argue that when these bands are partially filled, electron correlation could give rise to the quantum anomalous Hall (QAH) effect and the fractional QHE. Our results may open new directions focusing on topological phenomena in the rapidly growing field of oxide electronics.
Results
Design principle
To demonstrate the design principle for engineering TIs in TMO heterostructures, we consider perovskite-type TMOs as our prototype system. These compounds are very common and have the chemical formula ABO3, where O is oxygen and B is a transition-metal (TM) ion. The key idea is to start with a band structure that possesses 'Dirac points' in the Brillouin zone without the SOC, and then examine whether an energy gap can be opened at those points with the SOC turned on. If an energy gap does open, combined with proper filling the resulting state could be a TI. In an ideal perovskite structure, the TM ions sit on a simple cubic lattice, with the octahedral crystalline field splitting the TM d orbitals into twofold degenerate  and threefold degenerate
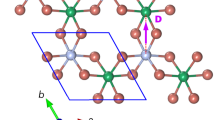
and threefold degenerate  levels, well separated by so-called 10Dq on the order of 3 eV. Such a lattice geometry usually does not support Dirac points. Instead, we consider bilayers of the perovskite structure grown in the [111] direction. As shown in Figure 1, the TM ions in the (111) bilayer are located on a honeycomb lattice consisting of two trigonal sublattices on different layers. This lattice geometry has three consequences: firstly, it is well known from the study of graphene that electrons hopping on a honeycomb lattice generally give rise to Dirac points in the band structure; secondly, a layer potential difference can be easily created by applying a perpendicular electric field or by sandwiching the bilayer between two different substrates, which allows experimental control of the band topology; and, thirdly, the honeycomb lattice further reduces the symmetry of the crystalline field from octahedral (Oh) to trigonal (C3v), and introduces additional level splitting of the d orbitals. The last point turns out to be crucial for realizing the topologically insulating phase.
levels, well separated by so-called 10Dq on the order of 3 eV. Such a lattice geometry usually does not support Dirac points. Instead, we consider bilayers of the perovskite structure grown in the [111] direction. As shown in Figure 1, the TM ions in the (111) bilayer are located on a honeycomb lattice consisting of two trigonal sublattices on different layers. This lattice geometry has three consequences: firstly, it is well known from the study of graphene that electrons hopping on a honeycomb lattice generally give rise to Dirac points in the band structure; secondly, a layer potential difference can be easily created by applying a perpendicular electric field or by sandwiching the bilayer between two different substrates, which allows experimental control of the band topology; and, thirdly, the honeycomb lattice further reduces the symmetry of the crystalline field from octahedral (Oh) to trigonal (C3v), and introduces additional level splitting of the d orbitals. The last point turns out to be crucial for realizing the topologically insulating phase.

(a) Perovskite structure ABO3. (b) A (111) bilayer consisting of the top layer indicated by red circles and the bottom layer indicated by blue circles. The lattice constant is a0. The bilayer shown as solid lines in (b) forms the honeycomb lattice when projected on the [111] plane with the lattice constant  (c). The real space coordinates are labelled by (x,y,z) in the original cubic lattice, while it is labelled by (X,Y) in the [111] plane. (d) Level structure of TM d orbital. In the cubic environment, d orbitals split into eg and t2g manifolds. With the SOC, t2g manifold further splits into two levels characterized by the effective total angular momentum j=1/2 and 3/2. With the trigonal crystal field, t2g manifold splits into two levels denoted by a1g and e′g. With both the SOC and the trigonal field, t2g manifold splits into three levels and eg manifold splits into two levels, that is, all the degeneracies are lifted except the Kramers doublets. (e) ABO3 monolayer is grown on AO3-terminated AB′O3 substrate capped by AB′O3. The direction of crystal growth is indicated by an arrow.
(c). The real space coordinates are labelled by (x,y,z) in the original cubic lattice, while it is labelled by (X,Y) in the [111] plane. (d) Level structure of TM d orbital. In the cubic environment, d orbitals split into eg and t2g manifolds. With the SOC, t2g manifold further splits into two levels characterized by the effective total angular momentum j=1/2 and 3/2. With the trigonal crystal field, t2g manifold splits into two levels denoted by a1g and e′g. With both the SOC and the trigonal field, t2g manifold splits into three levels and eg manifold splits into two levels, that is, all the degeneracies are lifted except the Kramers doublets. (e) ABO3 monolayer is grown on AO3-terminated AB′O3 substrate capped by AB′O3. The direction of crystal growth is indicated by an arrow.
We first consider the t2g manifold, in which the on-site SOC is active. In our modelling, only nearest-neighbour (NN) hopping of d electrons between the TM sites via oxygen p orbital is included. As we are interested in the band topology, which is robust against small perturbations as long as the band gap remains open, our model is justified and allows us to capture the essential ingredients with minimal parameterization. The TB Hamiltonian is given by

where r and τ label the lattice sites and the t2g orbitals, respectively. The first term is the hopping term represented by a single amplitude t and the dimensionless structural factor  . The second term is the on-site SOC, which splits the t2g levels into a j=1/2 doublet with energy λ and a j=3/2 quadruplet with energy −λ/2. lr and sr are the angular momentum and spin operators. The third term is the trigonal crystalline field which splits the t2g manifold into a1g and eg′ manifolds with their level separation given by 3Δ/2. V in the last term is the layer potential difference, and ξr=
. The second term is the on-site SOC, which splits the t2g levels into a j=1/2 doublet with energy λ and a j=3/2 quadruplet with energy −λ/2. lr and sr are the angular momentum and spin operators. The third term is the trigonal crystalline field which splits the t2g manifold into a1g and eg′ manifolds with their level separation given by 3Δ/2. V in the last term is the layer potential difference, and ξr= 1 when r is in the top or bottom layer. The explicit form of the Hamiltonian is presented in the Methods.
1 when r is in the top or bottom layer. The explicit form of the Hamiltonian is presented in the Methods.
The large number of orbitals (six per TM site) involved in our model give rise to a very rich behaviour of the topological band structure in the parameter space. Depending on the strength of the SOC, the system falls into two different phases. In the strong SOC limit (|λ/t|>8/3 when Δ=0), bands originating from the j=1/2 and j=3/2 orbitals are completely separated. The trigonal crystal field then opens up an energy gap within each manifold, and a non-trivial Z2 topology can be realized. This is similar to the results reported on Iridium compounds17,18. In the weak SOC limit, bands from j=1/2 and j=3/2 orbitals become mixed away from the Γ point. Again, we find topologically non-trivial energy gaps that can be opened by Δ. The Z2 topological invariant is determined using two different methods. One can either evaluate it directly from the bulk band structure19 or count the number of edge states. The calculated band structure for both cases together with the Z2 index is shown in Figure 2a–d. By inspection, we find that t2g1, t2g2 , t2g3 and t2g5 TMOs are all possible candidates for TIs in the strong SOC limit, and t2g2 , t2g4 and t2g5 in the weak SOC. The dependence of the band topology on the layer potential difference V is rather interesting. While increasing V will eventually destroy the Z2 non-trivial phase, under moderate values of V, the t2g3 system remains TI in the strong SOC limit, and the t2g4 system in the weak SOC limit.

(a) and (b) t2g model in the strong SOC limit. The SOC is fixed as λ/t=5 with Δ/t=1 (red) and Δ=0 (green). (c) and (d) t2g model in the weak SOC limit, Δ/t=0.5 with λ/t=1.5 (red), and Δ/t=1.5 with λ/t=0 (green). (e) and (f) eg model with  (red) and
(red) and  (green). Figures (a), (c) and (e) show the bulk dispersion relations. The dispersions in red correspond to the topologically non-trivial bands with the Z2 invariants shown for each band. Sum of Z2 in the occupied bands gives the Z2 topological invariant for the corresponding filling. For example, when the lowest five bands of the t2g model are occupied by electrons in (a), Z2 invariant becomes 1+0+0+1+1 mod 2=1. The insets in (a) and (c) show the zoom-up near the K point. Figures (b), (d) and (f) show the dispersion relations in finite-thick zigzag ribbons with the periodic boundary condition along the X direction and the openboundary condition along the Y direction. Parameters are the same as in the bulk dispersions. Edge modes supporting the spin current are indicated by red lines. For the t2g model with the weak SOC, there appear four edge channels between the third and the fourth bands as shown as blue lines in consistent with the Z2 number.
(green). Figures (a), (c) and (e) show the bulk dispersion relations. The dispersions in red correspond to the topologically non-trivial bands with the Z2 invariants shown for each band. Sum of Z2 in the occupied bands gives the Z2 topological invariant for the corresponding filling. For example, when the lowest five bands of the t2g model are occupied by electrons in (a), Z2 invariant becomes 1+0+0+1+1 mod 2=1. The insets in (a) and (c) show the zoom-up near the K point. Figures (b), (d) and (f) show the dispersion relations in finite-thick zigzag ribbons with the periodic boundary condition along the X direction and the openboundary condition along the Y direction. Parameters are the same as in the bulk dispersions. Edge modes supporting the spin current are indicated by red lines. For the t2g model with the weak SOC, there appear four edge channels between the third and the fourth bands as shown as blue lines in consistent with the Z2 number.
Next, we consider the eg manifold. It is well known that the SOC is quenched within the eg manifold so it seems that the resulting band topology should be trivial. However, similar to graphene20,21 and some TM ions22,23, the SOC can still take place through the virtual excitation of electrons between eg and t2g levels. According to the second-order perturbation theory, the effective SOC is given by

where ɛ labels the eg orbitals. Hence, the Hamiltonian can be written

where the SOC magnitude is given by  , with ΔE roughly being the energy difference between eg and t2g levels. (The explicit form of the Hamiltonian is presented in the Methods and in Supplementary Note 1.) We find that
, with ΔE roughly being the energy difference between eg and t2g levels. (The explicit form of the Hamiltonian is presented in the Methods and in Supplementary Note 1.) We find that  opens up an energy gap at the Γ point and also the Dirac point located at K. From the inspection of the Z2 topological invariant and counting the number of edge states, we found that eg1, eg2 and eg3 systems become TIs (Fig. 2e,f). Here, the trigonal crystalline field is also important—if all t2g levels are degenerated, even the second-order SOC vanishes. A layer potential difference comparable to
opens up an energy gap at the Γ point and also the Dirac point located at K. From the inspection of the Z2 topological invariant and counting the number of edge states, we found that eg1, eg2 and eg3 systems become TIs (Fig. 2e,f). Here, the trigonal crystalline field is also important—if all t2g levels are degenerated, even the second-order SOC vanishes. A layer potential difference comparable to  closes the gap at the Dirac point, turning the eg2 system into a trivial insulator. On the other hand, gaps at the Γ point are stable against this perturbation. Instead, these gaps close when the local potential difference between
closes the gap at the Dirac point, turning the eg2 system into a trivial insulator. On the other hand, gaps at the Γ point are stable against this perturbation. Instead, these gaps close when the local potential difference between  and
and  is comparable to
is comparable to  . Thus, the TI state and the Jahn–Teller effect24 compete in real materials with the eg1 or eg3 configuration.
. Thus, the TI state and the Jahn–Teller effect24 compete in real materials with the eg1 or eg3 configuration.
Materials consideration
Having established that the TIs can be realized in (111)-bilayer TMO for both t2g and eg configurations, we now turn to real materials. We aim to realize the integer fillings established above using TM B ions with the formal valence +3 or +4. For B3+(4+), we choose La (Sr) for the A-site element in both the target TMO and the insulating substrate AB′O3, and Al (Ti) for the B′-site element in the insulating substrate. Controlling the strain effects and the layer potential difference is possible by replacing A and/or B′ with their isovalent elements. It is well known that some of the TMOs are insulating due to strong correlations25. Therefore, if the corresponding bulk system is heavily insulating, bilayering may not be useful. Even if the corresponding bulk system is metallic, the low dimensionality in (111) bilayers may drive the system into a Mott insulator26. Further, the correlation effects are expected to reduce the effective band width and increase the splitting between occupied levels and unoccupied levels. While this effect does not change the band topology in eg electron systems, this could influence the topology in t2g systems by modifying the crystal field splitting between a1g and e′g levels. In addition, in a system with an integer number of electrons per site, local moments could be induced by the correlation effects resulting in the magnetic ordering. If the symmetry breaking by the magnetic ordering is strong, the system could become a trivial insulator. We do not consider such complexities by focusing on rather itinerant 4d and 5d electrons of TM ion, yet t2g electron systems are more susceptible for magnetic orderings than eg electron systems because of the smaller hopping intensity. These considerations somewhat limit the choice of TM and substrate material. Our candidate materials for TIs are, therefore, LaRe3+O3 as a t2g4 electron system, LaRu3+O3, LaOs3+O3, SrRh4+O3 and SrIr4+O3 as t2g5 systems, and LaAg3+O3 and LaAu3+O3 as eg2 electron systems. Most of these materials have been synthesized and their references are summarized in Table 1. LaReO3, LaOsO3 and LaAgO3 have yet to be synthesized. According to Ralle and Jansen35, bulk LaAuO3 has CaF2 structure rather than the perovskite. We expect this material shapes the perovskite structure by, for example, high-pressure synthesis and grown on a substrate with the perovskite structure. If properly synthesized, perovskite LaAuO3 is expected to be metallic as LaAgO3 predicted by the density functional theory (DFT) calculation33. We also note that growing thin films of perovskite TMOs along the [111] direction has already started36,37. While t2g2 systems are also candidates for TIs, the TI state is hard to realize because of the band overlap. For eg1 and eg3 systems, additional effects such as longer range transfer and the Jahn–Teller effect can easily modify the dispersion relations.
We first performed the DFT calculations for the bilayers of t2g systems LaReO3, LaRuO3, LaOsO3, SrRhO3 and SrIrO3 (details are presented in the Methods section.) Their dispersion relations are shown in Figure 3a–d. We notice the remarkable agreement between the DFT results and the TB result, Figure 2c, especially for LaReO3 and LaOsO3. For SrRhO3, LaReO3 and LaRuO3 (not shown), the Fermi level crosses several bands. Thus, these systems are classified as topological metals rather than TIs. In LaOsO3 and SrIrO3, the Fermi level is located inside the gap. Therefore, from the analogy to the TB model, (111) bilayers of LaOsO3 and SrIrO3 are TIs. From our DFT calculations, it is noted that the material dependence of the dispersion relations is rather large for t2g systems. This is because a large number of band parameters are involved in the band structure including the local crystalline field.

Symmetric bilayers: (a) LaReO3, (b) LaOsO3, (c) SrRhO3, (d) SrIrO3, (e) LaAgO3 and (f) LaAuO3. Bilayers shown in (a), (b), (e) and (f) are grown between LaAlO3, while those in (c) and (d) are grown between SrTiO3. Asymmetric bilayers of LaAuO3 grown between LaAlO3 and LaScO3 (g), and between LaAlO3 and YAlO3 (h). The Fermi level is taken to be 0 of the vertical axis. Bilayers shown in (b), (d), (e), (f) and (h) are TIs with the band gap indicated, (g) is a trivial insulator and others are topological metals.
We now move to eg2 electron systems, LaAgO3 and LaAuO3. Our DFT results for these systems are shown in Figure 3e,f. As in the t2g case, the DFT reproduces the TB result fairly well. In both undoped systems, the Fermi level is inside the gap at the K point, and from the analogy to the TB result, these systems are TIs. The gap amplitude is found to be about 150 meV for LaAuO3 and 40 meV for LaAgO3, so these systems should remain TIs at room temperature. The band gap and topological property can be controlled by breaking the symmetry between top and bottom layers. As shown in Figure 3g, the asymmetric bilayer with LaScO3 has larger band gap ~300 meV, and from the inspection of the symmetry of the wave function at the K point, this bilayer is a trivial insulator. On the other hand, the asymmetric bilayer with YAlO3 has smaller band gap ~50 meV and remains to be a TI (Fig. 3h). Such a small band gap TI is especially useful to control the topological property by using the gate voltage. Similar to LaAgO3 and LaAuO3, we also performed the DFT calculations for a 3d system LaCuO3. We found that this system develops an instability towards magnetic ordering because the itinerancy is reduced compared with 4d and 5d systems.
Nearly flat Z2 topologically non-trivial bands
One of the appealing aspect of realizing non-trivial band topology in TMOs is the rich possibilities of novel phenomena that could emerge when electron correlation is considered. Here, we demonstrate one of the possibilities in the eg systems with nearly flat Z2 topologically non-trivial bands (see Fig. 2e). Obviously, when the chemical potential is tuned into the nearly flat Z2 bands, we are in a novel regime of quantum many-body physics. From the inspection of dispersion relations shown in Figure 3e and f, the upper flat band appears to be more stable than the lower flat band. Tuning the chemical potential in the upper flat band corresponds to removing electrons from d10 systems or adding electrons to d9 systems. When such a situation is realized, kinetic energy is suppressed and physics is controlled mainly by interaction, whose energy scale is typically 1–2 eV, much larger than the width of flat bands W~0.2 eV. What would be the ground state of the system? Here, we propose several natural candidate states. One very likely consequence of the short-range repulsion U, when the doping of the nearly flat band is not too small, is to drive the system ferromagnetic because of Stoner's criteria U/W»1. On the other hand, when doping is too small, Wigner crystal phase should naturally occur. In the following, we assume that spontaneous ferromagnetic ordering occurs. The magnetic order should be viewed as a breaking of a discrete symmetry, due to the SOC, instead of the breaking of a continuous spin rotation symmetry, which is not realized in a 2D system at any finite temperature. Our discussion is not limited to spontaneous ordering, because magnetism can be also introduced externally by using a magnetic insulator as a substrate.
We model the effect of ferromagnetic ordering by adding a Zeeman spin splitting to the Hamiltonian:

Resulting dispersion relations for eg system with two characteristic Zeeman fields are presented in Figure 4. From the inspection of the Chern number, the QAH-insulating state38 is realized in eg0.5 and eg3.5 systems, and also eg1, eg1.5, eg2.5 and eg3 systems when the Zeeman field is large. The eg1 configuration with the large Zeeman splitting is realized in undoped perovskite manganites. More interesting physics occurs when the nearly flat 'Chern' band is partially filled (see Figs 2e and 4). In this case, as pointed out recently, fractional quantum Hall (FQH) states are likely to be realized39,40,41,42,43,44. To elaborate this possibility, we perform exact diagonalization calculation after projecting on-site repulsion and NN repulsion into the 1/3 filled highest Chern band (that is, at eg3.5+1/6). Indeed, signatures of a ν=1/3 FQH state are observed (details of the exact diagonalization and the numerical result are presented in the Supplementary Note 2). We find that, when the NN repulsive interaction is larger than the width of the flat band, the ground state degeneracy is threefold on a torus, and the Chern number (excluding the integer Chern number from the filled bands) of the ground state wave function is ~1/3 up to finite size correction.

(a) Small Zeeman splitting with B=0.3t, and (b) large Zeeman splitting with B=2t. Here, we used  . Bands with the majority (minority) spin component are indicated as red (blue) lines. Band-dependent Chern number is also indicated.
. Bands with the majority (minority) spin component are indicated as red (blue) lines. Band-dependent Chern number is also indicated.
The aforementioned FQH effects and QAH effects are both high-temperature effects and fundamentally different from the quantum Hall states realized in GaAs 2D electron gas in a magnetic field, where quasi-particle energy gap is controlled by the long-range Coulomb repulsion  (ɛ is the dielectric constant and lB is the magnetic length), typically around a few Kelvin. In the present systems, quasi-particle energy gap is determined by the short-range repulsion ~1–2 eV. This indicates that room temperature FQH effects may be realized. FQH states, in particular the non-Abelian states, have been shown to be very useful as building blocks of a quantum computer45,46. A high-temperature non-Abelian quantum Hall states in the TMO heterostructures, for example, at ν=1/2 filling where natural candidate states are in the same universality class of Pfaffian states47 or anti-Pfaffian states48,49, if realized experimentally, would have strong impacts on both fundamental physics and its applications, including the efforts of realizing topological quantum computation.
(ɛ is the dielectric constant and lB is the magnetic length), typically around a few Kelvin. In the present systems, quasi-particle energy gap is determined by the short-range repulsion ~1–2 eV. This indicates that room temperature FQH effects may be realized. FQH states, in particular the non-Abelian states, have been shown to be very useful as building blocks of a quantum computer45,46. A high-temperature non-Abelian quantum Hall states in the TMO heterostructures, for example, at ν=1/2 filling where natural candidate states are in the same universality class of Pfaffian states47 or anti-Pfaffian states48,49, if realized experimentally, would have strong impacts on both fundamental physics and its applications, including the efforts of realizing topological quantum computation.
Discussion
Before closing, we make few remarks on TIs in the TMO bilayers. The direct confirmation of the TI state is possible by measuring the conductance. As in Bernevig et al.5 and König et al.8, the conductance should be quantized as σ=2e2/h per (111) bilayer in the two-terminal measurement. The conductance can be controlled by using the gate voltage. In our DFT calculations, the (111) bilayers are repeated along the [111] axis. With the non-zero inter-bilayer coupling, the helical edge channels on the surface of the sample will turn to the two Dirac fermions at k[111]=0 and 1/2 in the unit of the reciprocal lattice vector along the [111] direction. Thus, strictly speaking, the TI is classified as a 'weak' TI and the backward scattering between the two Dirac fermions, if it exists, causes the localization. In order to avoid this, one needs to make the single (111) bilayer or keep the neighbouring (111) bilayers far apart so that the inter-bilayer coupling becomes exponentially small. When fabricating the (111) bilayer, a small number of defects would have minor effects because the edge modes surrounding them are disconnected. However, as the defect density increases, two surfaces are eventually connected through the edge modes belonging to the islands. As a result, the backward scattering takes place. Another source of the localization is islands of thicker (111) TMO layers. The suppressed trigonal field inside such thick islands is expected to create the gapless bulk modes. In further thicker islands of (111) TMO layers, metallic state would be realized inside the sample. In addition to (111) bilayers, we have studied model (111) trilayers and found that the TI states are robust without the even–odd oscillation between TI and trivial insulator, as predicted for the bismuth thin films50. Structurally, a (111) trilayer forms a so-called dice lattice, which could also bring about interesting quantum effects characterized by the Chern number C=±2 (ref. 51). Of course, if the layer structure is too thick then the bulk cubic symmetry is restored and the system is no longer a TI. So far, we did not mention the correlation effects and competing ground states except for the previous section. For limiting cases, we have performed unrestricted Hartree–Fock calculations for multi-orbital Hubbard models defined on (111) bilayers. We found that the TI states are rather robust for eg2 systems and become unstable against antiferromagnetic insulating states when the interaction strength is comparable to the full bandwidth as in the 2D Hubbard model on the honeycomb lattice52. For eg1 or eg3 systems, the QAH-insulating states could be generated dynamically by correlation effects without the SOC53,54, yet trivial-insulating states due to the Jahn–Teller effect would also be stabilized depending on the relative balance between the Coulomb interaction and the Jahn–Teller coupling. In this paper, we focused on the perovskite-type TMOs. Thus, our design principle for the TI state works only for the [111] plane because other planes such as [001] and [110] do not support a honeycomb lattice. However, this approach is not limited to the perovskite systems. For example, the [0001] plane of corundum Al2O3, that is, sapphire, involves a honeycomb lattice formed by Al atoms. Such a system could also be utilized as the substrate material to artificially create the TI state.
Methods
TB models in the real space
First, we consider a general multiband TB model on a cubic lattice given by

where r labels the TM sites, σ spin and μ orbitals.  is a transfer matrix, which depends on the pair of orbitals but not on the spin; its detail will be presented shortly.
is a transfer matrix, which depends on the pair of orbitals but not on the spin; its detail will be presented shortly.
For t2g electron systems, the trigonal crystal field directly couples with the local t2g level. In addition, the angular momentum is not quenched, and therefore the SOC is active. Including these two effects, a TB model for t2g systems is written as  with HSO and Htri given by the second and the third terms of equation (1), respectively. The explicit form of HSO for the t2g-alone model is given by
with HSO and Htri given by the second and the third terms of equation (1), respectively. The explicit form of HSO for the t2g-alone model is given by

with the use of the following convention for the orbital index:  ,
,  and
and  . στ with τ=a,b,c is the Pauli matrix, and
. στ with τ=a,b,c is the Pauli matrix, and  is the Levi–Civita antisymmetric tensor.
is the Levi–Civita antisymmetric tensor.
The dependence of transfer matrices on the orbital and direction is given by the Slater–Koster formula55 as follows:


for the NN hopping and



for the second-neighbour (SN) hopping. Here,  are the unit vector along the x, y and z direction, respectively. Although it is via weak π hybridization tpdπ between a TM ion and an oxygen ion, the NN hopping
are the unit vector along the x, y and z direction, respectively. Although it is via weak π hybridization tpdπ between a TM ion and an oxygen ion, the NN hopping  is the largest parameter in this model, thus, taken as the unit of energy t. Δpd is the level difference between TM d orbitals and oxygen p orbitals. The ratio between
is the largest parameter in this model, thus, taken as the unit of energy t. Δpd is the level difference between TM d orbitals and oxygen p orbitals. The ratio between  and tπ is the dimensionless parameter
and tπ is the dimensionless parameter  tδ′ is also the NN hopping, but it is via weak direct overlap and, therefore, is expected to be small. tσ′′ and tπ′ are the SN hoppings due to the higher-order processes involving the transfer between two oxygen ions as
tδ′ is also the NN hopping, but it is via weak direct overlap and, therefore, is expected to be small. tσ′′ and tπ′ are the SN hoppings due to the higher-order processes involving the transfer between two oxygen ions as  As
As  involves relatively strong (weak) σ(π) hybridization between two oxygen ions
involves relatively strong (weak) σ(π) hybridization between two oxygen ions  we expect
we expect  Typical transfer intensities are shown in Supplementary Fig. S1(a).
Typical transfer intensities are shown in Supplementary Fig. S1(a).
For eg electron systems, linear coupling with the trigonal crystal field is absent. Therefore, the C3 lattice symmetry of a (111) bilayer does not influence the on-site eg level. On the other hand, eg degeneracy can be lifted by the distortion of an O6 cage surrounding a TM ion, that is, the Jahn–Teller effect. Focusing on the metallic regime, we neglect this effect. The angular momentum is quenched unless the coupling between eg and t2g orbitals are considered. We also neglect this effect at the moment but reconsider it later. Thus, for the eg-alone model,  The dependence of transfer matrices on the orbital and direction is again given by the Slater–Koster formula55. For the NN hopping, we have
The dependence of transfer matrices on the orbital and direction is again given by the Slater–Koster formula55. For the NN hopping, we have



and for the SN hopping



Here, ɛ(=α, β) labels the eg orbitals as  and
and  For eg electron systems, the NN hopping
For eg electron systems, the NN hopping  is via strong σ hybridization between a TM ion and an oxygen ion tpdσ and, therefore, largest. This hopping integral is taken as the unit of energy t. Again, the dimensionless parameter
is via strong σ hybridization between a TM ion and an oxygen ion tpdσ and, therefore, largest. This hopping integral is taken as the unit of energy t. Again, the dimensionless parameter  is defined by the ratio between
is defined by the ratio between  and tσ as
and tσ as  The NN hopping tδ is due mainly to the direct overlap between two TM ions and, therefore, expected to be small
The NN hopping tδ is due mainly to the direct overlap between two TM ions and, therefore, expected to be small  as tδ′ in the t2g orbital model. tσ′ is the SN hopping due to the higher-order processes involving the transfer between two oxygen ions as
as tδ′ in the t2g orbital model. tσ′ is the SN hopping due to the higher-order processes involving the transfer between two oxygen ions as  . Typical transfer intensities are shown in Supplementary Fig. S1(b).
. Typical transfer intensities are shown in Supplementary Fig. S1(b).
TB models on the (111) bilayer
By constraining the atomic coordinate r within the (111) bilayer R=(X,Y), it is straightforward to derive the TB Hamiltonian as a function of 2D momentum k=(kx, ky). We use the convention in which the projection of the NN bond into the (111) plane  is taken as the unit of the length scale. This is a factor
is taken as the unit of the length scale. This is a factor  smaller than the lattice constant of the cubic perovskite, and the size of the new unit cell is
smaller than the lattice constant of the cubic perovskite, and the size of the new unit cell is  Taking the primitive lattice vectors as
Taking the primitive lattice vectors as  and
and  the first Brillouin zone is a hexagon with six corners located at
the first Brillouin zone is a hexagon with six corners located at 
For the t2g orbital model, we obtain

with









Here, the spin indices are suppressed for simplicity. ∓V/2 is the sublattice-dependent potential, which breaks the symmetry between the top (labelled 1) and bottom (labelled 2) layers.
For the eg orbital model, we obtain

with






DFT calculations
DFT calculations were carried out using the projector augmented wave method56 with the generalized gradient approximation in the parametrization of Perdew, Burke and Enzerhof57 for exchange correlation as implemented in the Vienna Ab Initio Simulation Package58. The default plane-wave energy cutoff for O, 400.0 eV, was consistently used in all the calculations. The optimized crystal parameter is 3.81 Å for bulk LaAlO3, and 3.95 Å for bulk SrTiO3. These values are in consistent with the experimental values of 3.79 Å (Berkstresser et al.59) and 3.91 Å (Hellwege and Hellwege60). The TM bilayer structures were simulated by a supercell consisting of 12 AO3 and 12 B layers along the [111] direction with (A,B)=(La,Al) or (Sr,Ti) with two adjacent B layers replaced by TM ions. In the (111) plane, the supercell contains a 1×1 unit cell. A 6×6×1 special k-point mesh including the Γ point (0,0,0) was used for integration over the Brillouin zone. Optimized atomic structures were achieved when forces on all the atoms were <0.01 eV/Å.
Additional information
How to cite this article: Xiao, D. et al. Interface engineering of quantum Hall effects in digital transition-metal oxide heterostructures. Nat. Commun. 2:596 doi: 10.1038/ncomms1602 (2011).
References
The Quantum Hall Effect edited by Prange R.E. and Girvin S.M. (Springer-Verlag, 1987).
Thouless, D. J., Kohmoto, M., Nightingale, M. P. & den Nijs, M. Quantized Hall conductance in a two-dimensional periodic potential. Phys. Rev. Lett. 49, 405–408 (1982).
Haldane, F. D. M. Model for a quantum Hall effect without Landau levels: condensed-matter realization of the 'Parity Anomaly'. Phys. Rev. Lett. 61, 2015–2018 (1988).
Kane, C. L. & Mele, E. J. z2 topological order and the quantum spin Hall effect. Phys. Rev. Lett. 95, 146802 (2005).
Bernevig, B. A., Hughes, T. L. & Zhang, S.- C. Quantum spin Hall effect and topological phase transition in HgTe quantum wells. Science 314, 1757–1761 (2006).
Moore, J. E. & Balents, L. Topological invariants of time-reversal-invariant band structures. Phys. Rev. B 75, 121306 (2007).
Fu, L., Kane, C. L. & Mele, E. J. Topological insulators in three dimensions. Phys. Rev. Lett. 98, 106803 (2007).
König, M. et al. Quantum spin Hall insulator state in HgTe quantum wells. Science 318, 766–770 (2007).
Hsieh, D. et al. A topological dirac insulator in a quantum spin Hall phase. Nature 452, 970–974 (2008).
Xia, Y. et al. Observation of a large-gap topological-insulator class with a single Dirac cone on the surface. Nature Phys. 5, 398–402 (2009).
Qi, X.- L., Hughes, T. L. & Zhang, S.- C. Topological field theory of time-reversal invariant insulators. Phys. Rev. B 78, 195424 (2008).
Fu, L. & Kane, C. L. Superconducting proximity effect and Majorana fermions at the surface of a topological insulator. Phys. Rev. Lett. 100, 096407 (2008).
Izumi, M. et al. Perovskite superlattices as tailored materials of correlated electrons. Mat. Sci. Eng. B 84, 53–57 (2001).
Ohtomo, A., Muller, D. A., Grazul, J. L. & Hwang, H. Y. Artificial charge-modulation in atomic-scale perovskite titanate superlattices. Nature 419, 378–380 (2002).
Ohtomo, A. & Hwang, H. Y. A high-mobility electron gas at the LaAlO3/SrTiO3 heterointerface. Nature 427, 423–426 (2004).
Mannhart, J. & Schlom, D. G. Oxide interfaces—an opportunity for electronics. Science 327, 1607–1611 (2010).
Shitade, A. et al. Quantum spin Hall effect in a transition metal oxide Na2IrO3 . Phys. Rev. Lett. 102, 256403 (2009).
Pesin, D. & Balents, L. Mott physics and band topology in materials with strong spin-orbit interaction. Nature Phys. 6, 376–381 (2010).
Fu, L. & Kane, C. L. Time reversal polarization and a z2 adiabatic spin pump. Phys. Rev. B 74, 195312 (2006).
Min, H. et al. Intrinsic and Rashba spin-orbit interactions in graphene sheets. Phys. Rev. B 74, 165310 (2006).
Yao, Y., Ye, F., Qi, X.- L., Zhang, S.- C. & Fang, Z. Spin-orbit gap of graphene: first-principles calculations. Phys. Rev. B 75, 041401 (2007).
Vallin, J. T. Dynamic Jahn-Teller effect in the orbital 2E state of Fe2+ in CdTe. Phys. Rev. B 2, 2390 (1970).
Chen, G., Balents, L. & Schnyder, A. P. Spin-orbit singlet and quantum critical point on the diamond lattice: FeSc2S4 . Phys. Rev. Lett. 102, 096406 (2009).
Jahn, H. A. & Teller, E. Stability of polyatomic molecules in degenerate electronic states. I. Orbital degeneracy. Proc. Roy. Soc. London 161, 220–235 (1937).
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039–1263 (1998).
Yoshimatsu, K. et al. Dimensional-crossover-driven metal-insulator transition in SrVO3 ultrathin films. Phys. Rev. Lett. 104, 147601 (2010).
Sugiyama, T. & Tsuda, N. Electrical and magnetic properties of Ca1−xLaxRuO3 . J. Phys. Soc. Jpn. 68, 3980–3987 (1999).
Yamaura, K. & Takayama-Muromachi, E. Enhanced paramagnetism of the 4d itinerant electrons in the rhodium oxide perovskite SrRhO3 . Phys. Rev. B 64, 224424 (2001).
Desu, S. B., Yoo, I. K., Kwok, C. K., & Vijay, D. P. Multilayer electrodes for ferroelectric devices European Patent EP0636271 (1995).
Cao, G. et al. Non-Fermi-liquid behavior in nearly ferromagnetic SrIrO3 single crystals. Phys. Rev. B 76, 100402(R) (2007).
Moon, S. J. et al. Dimensionality-controlled insulator-metal transition and correlated metallic state in 5d transition metal oxides Srn+1IrnO3n+1 (n=1,2, and ∞). Phys. Rev. Lett. 101, 226402 (2008).
Sumi, A. et al. MOCVD growth of epitaxial SrIrO3 films on (111)SrTiO3 substrates. Thin Solid Films 486, 182–185 (2005).
Bacalis, N. C. Band structure and electron-phonon interaction of LaAgO3 . J. Superconductivity 1, 175–180 (1988).
Guruswany, V., Keillor, P., Campbell, G. L. & Bockris, J.O'M. The photoelectrochemical response of the lanthanides of chromium, rhodium, vanadium and gold on a titanium base. Solar Energy Materials 4, 11–30 (1980).
Ralle, M. & Jansen, M. Synthesis and crystal structure determination of LaAuO3 . J. Solid State Chem. 105, 378–384 (1993).
Chakraverty, S., Ohtomo, A., Okude, M., Ueno, K. & Kawasaki, M. Epitaxial structure of (001)- and (111)-oriented perovskite ferrate films grown by pulsed-laser deposition. Cryst. Growth Des. 10, 1725–1729 (2010).
Gray, B., Lee, H.- Y., Liu, J., Chakhalian, J. & Freeland, J. W. Local electronic and magnetic studies of an artificial La2FeCrO6 double perovskite. Appl. Phys. Lett. 97, 013105 (2010).
Nagaosa, N. et al. Anomalous Hall effect. Rev. Mod. Phys. 82, 1539–1592 (2010).
Tang, E., Mei, J.- W. & Wen, X.- G. High-temperature fractional quantum Hall states. Phys. Rev. Lett. 106, 236802 (2011).
Sun, K., Gu, Z., Katsura, H. & Das Sarma, S. Nearly flatbands with nontrivial topology. Phys. Rev. Lett. 106, 236803 (2011).
Neupert, T., Santos, L., Chamon, C. & Mudry, C. Fractional quantum Hall states at zero magnetic field. Phys. Rev. Lett. 106, 236804 (2011).
Venderbos, J. W. F., Daghofer, M. & van den Brink, J. Narrowing of topological bands due to electronic orbital degrees of freedom. Phys. Rev. Lett. 107, 116401 (2011).
Wang, Y.- F., Gu, Z.- C., Gong, C.- D. & Sheng, D. N. Fractional quantum Hall effect of hard-core bosons in topological flat bands. Phys. Rev. Lett. 107, 146803 (2011).
Neupert, T., Santos, L., Ryu, S., Chamon, C. & Mudry, C. Fractional topological liquids with time-reversal symmetry and their lattice realization. Phys. Rev. B 84, 165107 (2011).
Bonderson, P., Kitaev, A. & Shtengel, K. Detecting non-abelian statistics in the ν=5/2 fractional quantum Hall state. Phys. Rev. Lett. 96, 016803 (2006).
Nayak, C., Simon, S. H., Stern, A., Freedman, M. & Das Sarma, S. Non-abelian anyons and topological quantum computation. Rev. Mod. Phys. 80, 1083–1159 (2008).
Moore, G. & Read, N. Nonabelions in the fractional quantum Hall effect. Nucl. Phys. B 360, 362–396 (1991).
Levin, M., Halperin, B. I. & Rosenow, B. Particle-hole symmetry and the Pfaffian state. Phys. Rev. Lett. 99, 236806 (2007).
Lee, S.- S., Ryu, S., Nayak, C. & Fisher, M. P. A. Particle-hole symmetry and the ν=5/2 quantum Hall state. Phys. Rev. Lett. 99, 236807 (2007).
Murakami, S. Quantum spin Hall effect and enhanced magnetic response by spin-orbit coupling. Phys. Rev. Lett. 97, 236805 (2006).
Wang, F. & Ran, Y. Nearly flat band with Chern number C=2 on the dice lattice. Preprint arXiv:1109.3435 (2011).
Meng, Z. Y., Lang, T. C., Wessel, S., Assaad, F. F. & Muramatsu, A. Quantum spin liquid emerging in two-dimensional correlated Dirac fermions. Nature 464, 847–852 (2010).
Rüegg, A. & Fiete, G. A. Topological insulators from complex orbital order in transition-metal oxides heterostructures. Phys. Rev. B 84, 201103(R) (2011).
Yang, K.- Y. et al. Possible interaction-driven topological phases in (111) bilayers of LaNiO3 . Phys. Rev. B 84, 201104(R) (2011).
Slater, J. C. & Koster, G. F. Simplified LCAO method for the periodic potential problem. Phys. Rev. 94, 1498–1524 (1954).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for Ab Initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Berkstresser, G. W., Valentino, A. J. & Brandle, C. D. Growth of single crystals of lanthanum aluminate. J. Cryst. Growth 109, 467–471 (1991).
Hellwege, K.- H. & Hellwege, A. M. (eds) Landolt-Börnstein: Numerical Data and Functional Relationships in Science and Technology New Series, Group III, Vol. 16a, 59–64 (Springer, 1981).
Acknowledgements
We thank H. Christen, K. Yamaura, Wanxiang Feng and A. Rüegg for their stimulating discussions. Work by W.Z. and S.O. was supported by the US Department of Energy, Office of Basic Energy Sciences, Materials Sciences and Engineering Division. D.X. was supported by the Laboratory Directed Research and Development Program of ORNL. N.N. acknowledges support from MEXT Grant-in-Aid for Scientific Research (19048008, 19048015 and 21244053). Computational support was provided by NERSC of US Department of Energy.
Author information
Authors and Affiliations
Contributions
S.O. conceived the idea for the (111) bilayer and constructed the theoretical models. Model calculations were performed by D.X. and S.O. (non-interacting models), and Y.R. (interacting models). The density functional theory calculations were performed by W.Z. N.N. provided further theoretical inputs. S.O. and D.X. were responsible for overall project direction, planning and management.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S2, Supplementary Notes 1-2 and Supplementary References (PDF 297 kb)
Rights and permissions
About this article
Cite this article
Xiao, D., Zhu, W., Ran, Y. et al. Interface engineering of quantum Hall effects in digital transition metal oxide heterostructures. Nat Commun 2, 596 (2011). https://doi.org/10.1038/ncomms1602
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms1602
This article is cited by
-
Signatures of fractional quantum anomalous Hall states in twisted MoTe2
Nature (2023)
-
Magnetism and berry phase manipulation in an emergent structure of perovskite ruthenate by (111) strain engineering
npj Quantum Materials (2023)
-
Flux-flow instability across Berezinskii Kosterlitz Thouless phase transition in KTaO3 (111) based superconductor
Communications Physics (2023)
-
High Chern numbers in a perovskite-derived dice lattice (LaXO3)3/(LaAlO3)3(111) with X = Ti, Mn and Co
Scientific Reports (2023)
-
Novel self-assembled two-dimensional layered oxide structure incorporated with Au nanoinclusions towards multifunctionalities
Nano Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
