
Abstract
Coral calcification is dependent on the mutualistic partnership between endosymbiotic zooxanthellae and the coral host. Here, using newly developed geochemical proxies (δ11B and B/Ca), we show that Porites corals from natural reef environments exhibit a close (r2 ∼0.9) antithetic relationship between dissolved inorganic carbon (DIC) and pH of the corals’ calcifying fluid (cf). The highest DICcf (∼ × 3.2 seawater) is found during summer, consistent with thermal/light enhancement of metabolically (zooxanthellae) derived carbon, while the highest pHcf (∼8.5) occurs in winter during periods of low DICcf (∼ × 2 seawater). These opposing changes in DICcf and pHcf are shown to maintain oversaturated but stable levels of carbonate saturation (Ωcf ∼ × 5 seawater), the key parameter controlling coral calcification. These findings are in marked contrast to artificial experiments and show that pHcf upregulation occurs largely independent of changes in seawater carbonate chemistry, and hence ocean acidification, but is highly vulnerable to thermally induced stress from global warming.
Similar content being viewed by others

Introduction
Scleractinian corals together with their endosymbiotic dinoflagellates, Symbiodinium (zooxanthellae), have been spectacularly successful in building the tropical coral reef edifices that dominate many shallow-water environments and harbour more than one-third of the oceans’ biodiversity. The ongoing viability of these iconic1 tropical reef systems is however in question2,3, with symbiont-bearing shallow-water corals now facing the combined challenge of both global warming and ocean acidification from rapidly rising levels of CO2 (ref. 4). Critical to the success of reef-building corals is their ability to extract dissolved inorganic carbon (DIC) from seawater and efficiently convert it into calcium carbonate, the major constituent of their skeletons. While much progress has been made in identifying many of the key elements of the biologic machinery that are integral to the biocalcification process5,6,7 (Fig. 1), there are still significant gaps in our understanding. Foremost is the relationship between declining seawater pH and its impact on pH upregulation of the coral’s extracellular calcifying fluid8,9,10, a process that occurs at least in part via Ca-ATPase pumping of Ca2+ ions into the calcifying region in exchange for the removal of protons11. Of equal but largely overlooked importance, are the mechanisms via which the various pH-dependent species of DIC (that is, CO2, HCO3− or CO32−) are produced, transported, and then inter-converted at the site of calcification. It has also long been recognized12,13 that light plays a key role in driving rates of calcification, and that light-enhanced calcification occurs as a result of the photosynthetic activity of endosymbiont dinoflagellates (zooxanthellae), providing both energy and additional carbon needed to drive calcification. The exact mechanism(s) by which coral calcification is linked to endosymbiont photosynthesis has, however, remained largely enigmatic at the polyp scale (Fig. 1) the zooxanthellae are physically separated from the site of calcification13,14,15 and, apart from pH, few direct measurements exist16 of the chemical conditions necessary to constrain the biocalcification process. Here we provide new evidence for an intimate link between the biologically mediated process of pHcf upregulation of the calcifying fluid and biological control over the concentration of DIC in the calcifying fluid (DICcf). We find that over annual timescales there is an inverse correlation between pHcf and DICcf. This acts to maintain relatively stable levels of aragonite saturation in the calcifying fluid, and hence near-optimal rates of coral calcification, despite large seasonally driven variations in metabolically supplied DIC.

Calcification occurs within the subcalicoblastic space from an initial seawater-derived fluid with additional metabolic sourced supply of DIC5,6,7. Elevation of calcifying fluid pHcf occurs via removal of protons from the calcification site by Ca2+-ATPase exchangers. The carbonic anhydrases (CA) catalyse the forward reactions converting CO2 into HCO3− ions7,34. Transfer of DIC into the subcalicoblastic space may occur via diffusion of CO2 and/or by HCO3− pumping via bicarbonate anion transporters (BAT)5,6,7. The link between the activity of zooxanthellae located in the oral coral endoderm tissue to the generation of metabolic DIC within the aboral endoderm and calicoblastic cells (orange) and transport to the calcifying fluid remains uncertain5,6,7 (Figure modified from McCulloch et al.31).
Results
Reef-water and coral calcifying fluid carbonate chemistry
To reconstruct the carbonate chemistry of the calcifying fluid from which corals precipitate their aragonite skeleton, we use the boron isotopic composition (δ11B) as a proxy for the calcifying fluid pHcf (refs 10, 17, 18). For determining the carbonate ion concentrations [CO32−]cf in the calcifying fluid, we use the combined δ11B-B/Ca proxy19. The application of the δ11B-B/Ca carbonate ion proxy has now been made possible by recent experimental measurements of the B/Ca carbonate ion distribution coefficient19, a major limitation of previous studies20 (see ‘Methods’ section). To examine how the chemistry of the calcifying fluid varies seasonally under ‘real-world’ reef conditions, we have analysed the skeletons of massive Porites collected from Davies Reef in the Great Barrier Reef and from Coral Bay Ningaloo Reef for which reef-water pH and sea-surface temperatures (SST) records are available21,22 (see ‘Methods’ section). Species of massive Porites coral are ideal for reconstructing seasonal changes in the composition of their calcifying fluid since they are long-lived and, more importantly, the architecture of their skeleton has a relatively straightforward chronology that facilitates well-constrained timing of their skeletal growth at seasonal resolution23. Given that only limited records of seasonal changes in local seawater carbonate chemistry are available22,24, these data are supplemented by model estimates24 of the reef-induced pH variability. The Great Barrier Reef and Ningaloo Reef sites (see ‘Methods’ section) have a typical seasonal range in temperature from ∼23 to 28 °C, as well as relatively narrow seasonal ranges in seawater pHsw (total scale) from ∼8.02 in summer to ∼8.08 in winter (Fig. 2). This limited seasonal range in average reef-water pHsw of ∼0.06 pH units is comparable to that observed in the open ocean25, a reflection of the tight balance between production and respiration24 combined with the limited residence time of waters in most wave and tidally driven reef systems21.

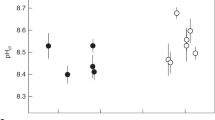
(a) Porites spp. coral calcifying fluid pHcf derived from δ11B systematics (see ‘Methods’ section and Supplementary Data) for colonies D-2 and D-3 from Davies Reef (18.8° S) in the Great Barrier Reef, Queensland. Shading denotes the summer period when pHcf and seawater pHsw values are at a minimum. Dashed line shows pH*cf expected from artificial experimental calibrations (pH*cf=0.32 pHsw+5.2)10,17 with an order of magnitude lower seasonal range than measured pHcf values. (b) Same as previous for Porites colonies from Coral Bay (CB-1 and CB-2) in the Ningaloo Reef of Western Australia (23.2° S) showing seasonal fluctuations in pHcf and seawater pHsw. The blue shading denotes the anomalously cool summer temperatures in 2010. (c) Enrichments in calcifying fluid DICcf (left axis; coloured circles) derived from combined B/Ca and δ11B systematics together with synchronous seasonal variations in reef-water temperatures (right axis; black line) for Porites colonies from Davies Reef (GBR). The strong temperature/light control on DICcf is consistent with enhanced metabolic activity of zooxanthellae symbionts in summer. (d) Same as previous but for Porites from Coral Bay (Ningaloo Reef, Western Australia).
Covariation of calcifying fluid pHcf and DICcf
In contrast to the limited variation in reef-water pHsw, we find that Porites colonies from both Davies Reef and Coral Bay exhibit strong seasonal changes in pHcf, from ∼8.3 during summer to ∼8.5 during winter (Fig. 2). This represents an elevation in pHcf relative to ambient seawater of ∼0.4 pH units together with a relatively large seasonal range in pHcf of ∼0.2 units. These observations are in stark contrast to the far more muted changes based on laboratory-controlled experiments9,17. These inferred laboratory responses10 in calcifying fluid pH (pH*cf) are shown in Figs 2 and 4, where the expected seasonal range is ∼0.02 pH units, an order of magnitude smaller than those actually observed in reef environments. The explanation for this unexpectedly large range in seasonal pHcf present under natural reef conditions becomes apparent from the exceptionally strong and inverse correlations between pHcf and DICcf (r2=0.88–0.94) present at the colony level (Fig. 3a,b).

(a) Covariation of calcifying fluid saturation state (Ωcf) with reef-water temperature showing a five- to sixfold elevation in Ωcf relative to reef-waters for Porites corals from Davies Reef (D-2 and D-3) in the Great Barrier Reef and from Coral Bay (CB-1 and CB-2) in the Ningaloo Reef. Note the very narrow range (±5 to ±10%) of high Ωcf values for each colony. (b) Subparallel arrays of inversely correlated (r2=0.88–0.94) calcifying fluid pHcf and DICcf/DICsw values reflecting specific bio-environmental controls at the colony level on metabolic DICcf/DICsw. Seasonal variations in metabolic supplied DICcf are offset by opposing changes in pHcf that act to moderate the overall variations in Ωcf, the ultimate controller of skeletal growth rates.

(a) Calcifying fluid pHcf and Ωcf values for Porites coral (D-2) from Davies Reef (GBR), where Ωcf=[Ca2+]cf [CO32−]cf/Karag. Dashed line shows the Ω*cf calculated using fixed experimental10,17 pH*cf values (see Fig. 2a,b). (b) Same as previous for Coral Bay (Ningaloo Reef, Western Australia) Porites (CB-2). (c) Calcification rates calculated using the inorganic rate equation28 G=k(Ω−1)n, where k and n are the temperature-dependent constant and order of the reaction, respectively28. Because of opposing changes in pHcf relative to DICcf (Fig. 1), Ωcf and hence coral growth rates are strongly modulated reducing seasonal variations by twofold compared to those estimated from fixed condition experiments (G*). (d) Same as previous for Porites from Coral Bay (Ningaloo Reef, Western Australia).
Here DICcf reaches its highest values in summer (× 2.0 to × 3.2 higher than ambient seawater) and lowest values in winter, whereas pHcf shows the opposite pattern. This seasonal variability in DICcf is consistent with light- and temperature-driven changes in the supply of metabolic DIC provided by the endosymbionts and/or the bicarbonate anion transporters within the coral host7. Thus, each Porites colony forms a distinctive subparallel array characterized by a distinctive range in DICcf that is inversely correlated to pHcf. Since the concentrations of carbonate ion [CO32−] and consequently the aragonite saturation state (Ωcf=[Ca2+]cf [CO32−]cf/Karag) of the calcifying fluid increases with increasing DICcf and pHcf, the observed antithetic seasonal changes in these parameters results in a more muted seasonal variation in Ωcf (±5% to ±10%, Fig. 3a) compared to that expected from changes in only pHcf (±30%) or DICcf (±12%) acting alone. While there remains a subdued positive correlation of Ωcf with temperature (Fig. 3a), the inverse correlations between pHcf and DICcf (Fig. 2a,b) indicate that the coral is actively maintaining both high (∼ × 4 to × 6 seawater) and relatively stable (within ±10% of mean) levels of elevated Ωcf year-round.
While the absolute levels of enhanced Ωcf are not dissimilar to previous qualitative estimates17,26, the finding of significantly higher but relatively limited ranges in DICcf of ∼ × 2.0 to ∼ × 3.2 seawater, is not generally consistent with recent micro-sensor16 measurements. This difference may reflect the intrinsic limitations6 of using probes that are 15–20 μm wide to measure the chemistry within the much narrower and irregular (1–10 μm) calcifying region. Additionally, separate probes are required for measurements of pHcf and [CO32−]cf, introducing further uncertainty, likely accounting for the large variability of in situ measured CO32− and hence inferred DICcf (∼ × 1.4 to × 4.2 seawater). Finally and most importantly, regardless of the method employed, we find that measurements conducted under controlled, static, laboratory conditions10 are unlikely to be representative of natural reef conditions due to the interactive dynamics of pHcf and DICcf upregulation described herein.
Discussion
The underlying reason for the dynamic, antiphase relationship between pHcf versus DICcf, can be explained by the ability of the coral to ‘control’ what is arguably27 one of its most fundamental physiological processes, the growth of its skeleton within which it lives. For example, during winter (Fig. 2), there is a large systematic decrease in the abundance of metabolic DIC (∼25%), presumably as a consequence of reductions in both light and temperature. Since higher pH shifts the carbonate equilibria to favour CO32− relative to HCO3−, the greater increase in pHcf in winter (∼8.5) compared to summer (∼8.3) increases the concentration of carbonate ions within the calcifying fluid (and therefore Ωcf) for the same DICcf. This increase in winter pHcf therefore partially counters the seasonal slowdown in host-symbiont carbon metabolism. Hence during the cooler periods, higher pHcf enhances Ωcf and hence partially mitigates the reduced temperature-dependent kinetics of calcification because rates of mineral precipitation are proportional to (Ω−1)n, where n is the temperature-dependent order of the reaction28 (n=1.3–2.0 for most reef habitats). During summer, the opposite behaviour is observed, with higher rates of metabolic DICcf partially offset by decreases in pHcf, resulting in a concomitant decrease in the carbonate saturation state of the calcifying fluid (Ωcf) and hence moderated (albeit still high) rates of calcification (Fig. 4c,d).
This implies that during summer, zooxanthellae-derived DICcf is being supplied in excess of the ‘optimal’ requirements for the biologically mediated process of skeleton building. Thus, while existing mineral rate kinetics indicate that rates of calcification are still a factor of two- to fourfold higher in summer than in winter, this range is significantly less than the estimated eightfold higher summer rates (Fig. 4c,d) if constant levels of elevated pHcf upregulation were operative, as implied from the artificial constant seawater pHsw and temperature experiments10.
Although our findings are based only on species of Porites from the Pacific and Indian Oceans, they nevertheless have important implications for our understanding of how reef-building corals in general will respond to climate change. The occurrence, for example, of the highest pHcf values during winter, when metabolically derived sources of energy are at a minimum, provides further evidence against the proposition that pHcf upregulation is an energetically costly process29, and will therefore decline as seawater pHsw decreases due to ocean acidification. This is supported by results of the free ocean carbon enrichment experiment30 conducted within the GBR Heron Island lagoon, where corals subjected to both natural and superimposed fluctuations in seawater pHsw exhibited essentially constant pHcf upregulation, a condition referred to by those authors30 as ‘pH homoeostasis’. These findings, combined with measurements of even higher pHcf in azooxanthellate deep-sea corals31 (pHcf >8.6), are thus consistent with inferences that Ca-ATPase-driven pHcf upregulation is a relatively energetically inexpensive process17. These observations, in conjunction with the highly correlated and anti-cyclical seasonal changes in both pHcf and DICcf, therefore argue against the reduction of pHcf in summer being a result of the passive feedback from higher rates of calcification producing more protons thereby lowering pHcf. Thus, while this possibility cannot yet be entirely excluded, the higher production rates of zooxanthellae-derived metabolites that are presumably available in the summer to facilitate enhanced Ca-ATPase activity, also suggest that the lower summer levels of pHcf is not due to intrinsic limitations in the Ca-ATPase H+ pumping, but rather physiological controls on growth rate. Furthermore, similar anti-correlated changes in pHcf and DICcf are present in Porites from both Davies and Ningaloo Reefs, despite large differences in growth rates.
Our findings also have major ramifications for the interpretation of the large number of experiments that have reported a strong sensitivity of coral calcification to increasing ocean acidification32. An inherent limitation of many of these experiments33 is that they were generally conducted under conditions of fixed seawater pHsw and/or temperature, light, nutrients, and little water motion, hence conditions that are not conducive to reproducing the natural interactive effects between pHcf and DICcf that we have documented here. A characteristic common to a variety of coral species grown under these artificial conditions is the apparently constant but limited sensitivity (one-third to one-half) of pHcf relative to external changes in seawater pHsw (refs 10, 17). While the reason for this apparently systematic but muted experimental response of pHcf is still uncertain, it likely involves reduced and/or constant levels of metabolically produced DICcf. Under such fixed conditions, we surmise that the supply of seawater DIC into the subcalicoblastic space (Fig. 1) becomes the dominant source and hence major influence on levels of DICcf, with upregulation of pHcf therefore acting as the major controller of Ωcf and thereby affecting the perceived sensitivity of pHcf to ocean acidification. This inference is supported by the fact that the observed pHcf of Porites from both Davies and Ningaloo Reefs were closest to the pHcf predicted from the constant condition experiments in winter when DICcf levels are naturally lowest due to reduced light and/or temperature, hence most similar to experimental predicted seawater end-member values. Clearly, since the interactive dynamics of pHcf and DICcf upregulation do not appear to be properly simulated under the short-term conditions generally imposed by such artificial experiments, the relevance of their commonly reported finding of reduced coral calcification with reduced seawater pH must now be questioned.
In summary, we have now identified the key functional characteristics of chemically controlled calcification in reef-building coral. The seasonally varying supply of summer-enhanced metabolic DICcf is accompanied by dynamic out-of-phase upregulation of coral pHcf. These parameters acting together maintain elevated but near-constant levels of carbonate saturation state (Ωcf) of the coral’s calcifying fluid, the key driver of calcification. Although the maintenance of elevated but near-constant Ωcf in mature coral colonies is not directly influenced by ocean acidification, it is however highly susceptible to thermal stress. In extreme cases of coral bleaching, the loss of endosymbionts disrupts the metabolic supply of DICcf as well as the metabolites necessary to operate the Ca-ATPase that upregulate pHcf (refs 14, 34), thus effectively terminating calcification. So, although rising levels of pCO2 can have adverse effects on the recruitment and growth of juvenile corals35,36,37,38, especially those lacking robust internal carbonate chemistry regulatory mechanisms, extreme thermal stress is detrimental to all symbiont-bearing corals39,40 regardless of their growth stage. We therefore conclude that the increasing frequency and intensity of coral bleaching events due to CO2-driven global warming constitutes the greatest immediate threat to the growth of shallow-water reef-building corals, rather than the closely associated process of ocean acidification.
Methods
Reef sites
Porites colonies were sampled from two reef systems: (1) Davies Reef (18.8° S, 147.63° E), a mid-shelf reef ∼100 km east-northeast of Townsville, Queensland, Australia in the central Great Barrier Reef, and (2) Coral Bay (23.19° S, 113.77° E), part of the Ningaloo Reef coastal fringing system of Western Australia. At Davies Reef, the annual range of daily average SST is 23–28.5 °C with a diurnal range of ∼0.5 °C or less41. In situ seawater temperature data extending back to 1987 for the core site at Davies Reef (18.83° S, 147.63° E) was compiled from a number of different temperature sensors deployed between a depth of ∼2 to ∼10 m maintained by the Australian Institute of Marine Science from October 1991 to December 2013 (http://data.aims.gov.au/aimsrtds/datatool.xhtml). To estimate seasonal changes in carbonate chemistry, we used the 24-h seawater carbonate chemistry data collected by Albright et al.22 on the lagoon side of the Davies Reef flat around the summer and winter extremes in both light and temperature. Their data showed that the daily average pH at that reef site was 8.02 in summer and 8.08 in winter; a seasonal range that was similar to seasonal minima and maxima observed and hind-cast at Coral Bay and hence similar to what would be expected from seasonal variations in temperature-driven pCO2 solubility. We therefore assumed that daily average pH at Davies Reef also followed seasonal changes in temperature according to pHsw=−0.010 × T+8.31.
At Coral Bay, SST generally ranges from 22–23 °C in winter to 27–28 °C in summer21. To hind-cast seasonal changes in reef-water temperature and pH, we first used time series of SST data from just offshore Coral Bay at ∼25 km resolution produced by Reynolds et al.42 before June 2010 and then at ∼1 km resolution produced by Chao et al.43 Both SST data products were then calibrated against in situ observations of temperature collected from a moored depth of ∼17 m as described by Falter et al.21 and previous model studies of wave-driven circulation. The carbonate chemistry of Coral Bay and offshore waters (∼2 km) were monitored between May 2011 and June 2012 and intermittently since then, with seasonal changes in offshore seawater pHT (total scale) being found to be strongly correlated with seasonal changes in offshore temperature (pHsw=−0.012 × T+8.37, r2=0.86, n=13). To determine seasonal changes in pH at the back-reef site where the coral cores were recovered, the offshore pH was adjusted to account for the deviation in temperature due to local heating and cooling (see above), as well as the daily average decrease in total alkalinity of ∼10 μmol kg−1 at back-reef sites observed from measurements44.
Boron isotopic pH proxy
Changes in the isotopic ratio of 11B (∼80%) and 10B (∼20%) are expressed in delta notation (in per mil, ‰) as:

where 11B/10Bcarb is the isotopic ratio measured in the coral carbonate and 11B/10BNIST951 is the isotopic ratio of the NIST SRM 951 boric acid standard. In seawater, boron exists as two different species, boric acid (B(OH)3) and the borate ion (B(OH)4−), with their relative abundance being pH dependent. The sensitivity of the δ11B proxy to the calcifying fluid pHcf arises from the incorporation of only the borate ion species into the aragonite structure45,46,47, with the δ11B isotopic composition reflecting the pH sensitivity of the borate versus boric acid speciation. The pH of the calcifying fluid (pHcf) can thus be calculated from the δ11B measured in the coral carbonate (δ11Bcarb). The equation used to convert the δ11Bcarb isotopic composition measured in the coral carbonate skeleton to a pH of the calcifying fluid (pHcf) is given by48:

where δ11Bsw represents the δ11B in seawater (δ11Bsw=39.61‰)49 and α(B3−B4)=1.0272 (ref. 50). The dissociation constant of boric acid pKB has a well-established value of 8.597 at 25 °C and a salinity of 35 (ref. 51). Here we also assume that the calcifying fluid has the same δ11B composition as seawater since that is the ultimate source of boron and, due to the low KD of B/Ca (ref. 19), the boron composition and concentration of the calcifying fluid remains essentially constant during calcification. Recent studies utilizing the δ11B pHcf proxy as well as direct measurements of calcifying fluid pH using pH-sensitive dyes9,18, have also confirmed that under highly controlled artificial conditions of constant pH and temperature, corals upregulate the pHcf of their calcifying fluid by 1/3 to ½ relative to ambient seawater pH.
B/Ca constraints on calcifying fluid DIC concentrations
Prior studies indicate that borate rather than boric acid is the predominant species occupying the lattice position normally taken up by the carbonate ion52 in calcifiers that precipitate aragonite skeletons. Although there are a number of reaction pathways through which this substitution could occur19,20, it is likely to involve de-protonation of the borate species to create a divalent base ion with the same charge as that of the carbonate ion species (−2), to preserve the charge neutrality of the growing crystal:

The partitioning of borate versus carbonate into aragonite is thus likely to be sensitive to solution pH10,19,20. Here the relevant partition coefficient KD relating the molar ratio of  to the concentrations of the carbonate
to the concentrations of the carbonate  and borate
and borate  species in the precipitating solution is determined using:
species in the precipitating solution is determined using:

Holcomb et al.19 conducted experiments quantifying the ratio of boron to calcium in aragonite precipitated inorganically under a wide range of carbonate chemistries (including pH) and total DIC and boron concentrations, as well as conditions of pH and DIC appropriate to those in the calcifying fluid of corals. Furthermore, Holcomb et al.19 also showed the close relationships between B/Ca, CO32− and KD based on substitution reactions between B(OH)4− and CO32−. Re-analysing the Holcomb et al.19 data, we find (Fig. 5) that the observed KD as defined in equation (4) shows the expected decrease as a function of the concentration of total active protons within the precipitating solution.

Measured B/Ca partition coefficient (KD) as defined by equation (4) from the data of Holcomb et al.19 The line represents a best-fit exponential curve to the data with KDB/Ca=KD,0 exp( [H+]T), where KD,0=2.97±0.17 × 10−3 (±95% CI),
[H+]T), where KD,0=2.97±0.17 × 10−3 (±95% CI),  =0.0202±0.042, r2=0.64 and n=63. The range for pHcf of upregulating calcifiers (that is, Porites spp.) is between ∼8 and ∼9 (shaded); equivalent to [H+]T of between 1 and 10 nmol kg−1 giving a range in KDB/Ca (× 10−3) of 2.6–2.8, and therefore relatively in-sensitive to changes in coral pHcf. Importantly, our experimentally determined KDB/Ca value is an order of magnitude higher than the previous estimate by Allison et al.,20 (open diamond) and consistent with the substitution of B(OH)4− with CO32−.
=0.0202±0.042, r2=0.64 and n=63. The range for pHcf of upregulating calcifiers (that is, Porites spp.) is between ∼8 and ∼9 (shaded); equivalent to [H+]T of between 1 and 10 nmol kg−1 giving a range in KDB/Ca (× 10−3) of 2.6–2.8, and therefore relatively in-sensitive to changes in coral pHcf. Importantly, our experimentally determined KDB/Ca value is an order of magnitude higher than the previous estimate by Allison et al.,20 (open diamond) and consistent with the substitution of B(OH)4− with CO32−.
Thus, using the definition of KD from equation (4) and its dependency on pHcf as given by the inorganic data of Holcomb et al.19, we can now calculate the concentration of carbonate ions within the calcifying fluid (that is, [CO32−]cf from measurements of (B/Ca)carb and pHcf, the latter derived from the skeletal boron isotopic ratio (δ11Bcarb). We further assume that [BT]cf is equal to the total concentration of boron of ambient seawater and only a function of seawater salinity ([BT]cf=[BT]sw at salinity=35). We therefore have:

Where KD=0.00297exp(−0.0202 [H+]T and for typical calcifying fluid pHcf values KD ∼0.0027, an order of magnitude higher than a previous estimate20. The concentration of DIC within the calcifying fluid is then calculated from the measured pHcf (equation 1) and  (equation 2) values using the programme CO2SYS provided by Lewis and Wallace53, with the carbonate species dissociation constants of Mehrbach et al.54 as re-fitted by Dickson and Millero55, the borate and sulfate dissociation constants of Dickson51,56, and the aragonite solubility constants of Mucci57. We also note that our use of a reliable experimentally determined KD is now consistent with substitution of borate with carbonate ion, rather than the previously inferred20 substitution with bicarbonate ion, the latter assumption effectively negating the role of carbonate saturation state on calcification.
(equation 2) values using the programme CO2SYS provided by Lewis and Wallace53, with the carbonate species dissociation constants of Mehrbach et al.54 as re-fitted by Dickson and Millero55, the borate and sulfate dissociation constants of Dickson51,56, and the aragonite solubility constants of Mucci57. We also note that our use of a reliable experimentally determined KD is now consistent with substitution of borate with carbonate ion, rather than the previously inferred20 substitution with bicarbonate ion, the latter assumption effectively negating the role of carbonate saturation state on calcification.
Data availability
The coral geochemical and seawater carbonate chemistry and temperature data are available in Supplementary Data.
Additional information
How to cite this article: McCulloch, M. T. et al. Coral calcification in a changing World and the interactive dynamics of pH and DIC upregulation. Nat. Commun. 8, 15686 doi: 10.1038/ncomms15686 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Darwin, C. The Voyage of the Beagle: Journal of Researches into the Natural History and Geology of the Countries Visited During the Voyage of HMS Beagle Round the World Modern Library (2010).
Hughes, T. P. et al. Climate change, human impacts, and the resilience of coral reefs. Science 301, 929–933 (2003).
Hoegh-Guldberg, O. Climate change, coral bleaching and the future of the world’s coral reefs. Mar. Freshw. Res. 50, 839–866 (1999).
Ciais, P. et al. in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (eds Stocker, T. F. et al.) 465–570 (Cambridge University Press, 2014).
Furla, P., Galgani, I., Durand, I. & Allemand, D. Sources and mechanisms of inorganic carbon transport for coral calcification and photosynthesis. J. Exp. Biol. 203, 3445–3457 (2000).
Allemand, D., Tambutté, E., Zoccola, D. & Tambutté, S. in Coral Reefs: An Ecosystem In Transition Vol. III (eds Dubinsky, Z. & Stambler, N.) 119–150 (Springer, 2011).
Zoccola, D. et al. Bicarbonate transporters in corals point towards a key step in the evolution of cnidarian calcification. Sci. Rep. 5, 9983 (2015).
Al-Horani, F. A., Al-Moghrabi, S. M. & de Beer, D. The mechanism of calcification and its relation to photosynthesis and respiration in the scleractinian coral Galaxea fascicularis. Mar. Biol. 142, 419–426 (2003).
Venn, A. A. et al. Impact of seawater acidification on pH at the tissue-skeleton interface and calcification in reef corals. Proc. Natl Acad. Sci. USA 110, 1634–1639 (2013).
Trotter, J. A. et al. Quantifying the pH ‘vital effect’ in the temperate zooxanthellate coral Cladocora caespitosa: validation of the boron seawater pH proxy. Earth Planet. Sci. Lett. 303, 163–173 (2011).
Cohen, A. L. & McConnaughey, T. in Biomineralization. Reviews in Mineralogy & Geochemistry, Vol. 54 (eds Dove, P. M., Weiner, S. & Yoreo, J. J.) Ch. 6, 151–187 (The Mineralogical Society of America, 2003).
Yonge, C. M., Nicholls, A. G. & Yonge, M. J. in Studies on the Physiology of Corals, Vol. 1 (British Museum, 1931).
Goreau, T. Coral skeletal chemistry: physiological and environmental regulation of stable isotopes and trace metals in Montastrea annularis. Proc. R. Soc. Lond. B Biol. Sci. 196, 291–315 (1977).
Allemand, D. et al. Biomineralisation in reef-building corals: from molecular mechanisms to environmental control. C. R. Palevol. 3, 453–467 (2004).
Tambutté, S. et al. Coral biomineralization: from the gene to the environment. J. Exp. Mar. Biol. Ecol. 408, 58–78 (2011).
Cai, W.-J. et al. Microelectrode characterization of coral daytime interior pH and carbonate chemistry. Nat. Commun. 7, 11144 (2016).
McCulloch, M. T., Trotter, J. A., Falter, J. & Montagna, P. Coral resilience to ocean acidification and global warming through pH up-regulation. Nat. Clim. Chang. 2, 623–627 (2012).
Holcomb, M. et al. Coral calcifying fluid pH dictates response to ocean acidification. Sci. Rep. 4, 5207–5211 (2014).
Holcomb, M., DeCarlo, T. M., Gaetani, G. A. & McCulloch, M. Factors affecting B/Ca ratios in synthetic aragonite. Chem. Geol. 437, 67–76 (2016).
Allison, N., Cohen, I., Finch, A. A., Erez, J. & Tudhope, A. W. Corals concentrate dissolved inorganic carbon to facilitate calcification. Nat. Commun. 5, 5741 (2014).
Falter, J. L. et al. Assessing the drivers of spatial variation in thermal forcing across a nearshore reef system and implications for coral bleaching. Limnol. Oceanogr. 59, 1241–1255 (2014).
Albright, R., Langdon, C. & Anthony, K. Dynamics of seawater carbonate chemistry, production, and calcification of a coral reef flat, central Great Barrier Reef. Biogeosciences 10, 6747–6758 (2013).
McCulloch, M. T., Gagan, M. K., Mortimer, G. E., Chivas, A. R. & Isdale, P. J. A high-resolution Sr/Ca and δ18O coral record from the Great Barrier Reef, Australia, and the 1982-1983 El Niño. Geochim. Cosmochim. Acta 58, 2747–2754 (1994).
Falter, J. L., Lowe, R. J., Zhang, Z. L. & McCulloch, M. Physical and Biological controls on the carbonate chemistry of coral reef waters: effects of metabolism, wave forcing, sea level, and geomorphology. PLoS ONE 8, e53303 (2013).
Takahashi, T. et al. Climatological distributions of pH, pCO2, total CO2, alkalinity, and CaCO3 saturation in the global surface ocean, and temporal changes at selected locations. Mar. Chem. 164, 95–125 (2014).
Holcomb, M., Cohen, A. L., Gabitov, R. I. & Hutter, J. L. Compositional and morphological features of aragonite precipitated experimentally from seawater and biogenically by corals. Geochim. Cosmochim. Acta 73, 4166–4179 (2009).
Fine, M. & Tchernov, D. Scleractinian coral species survive and recover from decalcification. Science 315, 1811 (2007).
Burton, E. A. & Walter, L. M. Relative precipitation rates of aragonite and Mg calcite from seawater: Temperature or carbonate ion control? Geology 15, 111–114 (1987).
Erez, J., Reynaud, S., Silverman, J., Scheinder, K. & Allemand, D. in Coral Reefs: an ecosystem in transition (eds Dubinsky, Z. & Stambler, N.) 151–176 (Springer, 2011).
Georgiou, L. et al. pH homeostasis during coral calcification in a free ocean CO2 enrichment (FOCE) experiment, Heron Island reef flat, Great Barrier Reef. Proc. Natl Acad. Sci. 112, 13219–13224 (2015).
McCulloch, M. T. et al. Resilience of cold-water scleractinian corals to ocean acidification: Boron isotopic systematics of pH and saturation state up-regulation. Geochim. Cosmochim. Acta 87, 21–34 (2012).
Chan, N. & Connolly, S. R. Sensitivity of coral calcification to ocean acidification: a meta‐analysis. Glob. Chang. Biol. 19, 282–290 (2013).
Gattuso, J. P. et al. Free-ocean CO2 enrichment (FOCE) systems: present status and future developments. Biogeosciences 11, 4057–4075 (2014).
Moya, A. et al. Carbonic anhydrase in the scleractinian coral Stylophora pistillata characterization, localization, and role in biomineralization. J. Biol. Chem. 283, 25475–25484 (2008).
Tambutté, E. et al. Morphological plasticity of the coral skeleton under CO2-driven seawater acidification. Nat. Commun. 6, 7368 (2015).
Foster, T., Falter, J. L., McCulloch, M. T. & Clode, P. L. Ocean acidification causes structural deformities in juvenile coral skeletons. Sci. Adv. 2, e1501130 (2016).
de Putron, S. J., McCorkle, D. C., Cohen, A. L. & Dillon, A. The impact of seawater saturation state and bicarbonate ion concentration on calcification by new recruits of two Atlantic corals. Coral Reefs 30, 321–328 (2011).
Albright, R. & Langdon, C. Ocean acidification impacts multiple early life history processes of the Caribbean coral Porites astreoides. Glob. Chang. Biol. 17, 2478–2487 (2011).
Randall, C. J. & Szmant, A. M. Elevated temperature affects development, survivorship, and settlement of the elkhorn coral, Acropora palmata (Lamarck 1816). Biol. Bull. 217, 269–282 (2009).
Chua, C. M., Leggat, W., Moya, A. & Baird, A. H. Temperature affects the early life history stages of corals more than near future ocean acidification. Mar. Ecol. Prog. Ser. 475, 85–92 (2013).
Alibert, C. & McCulloch, M. T. Strontium/calcium ratios in modern Porites corals from the Great Barrier Reef as a proxy for sea surface temperature: Calibration of the thermometer and monitoring of ENSO. Paleoceanography 12, 345–363 (1997).
Reynolds, R. W. et al. Daily high-resolution-blended analyses for sea surface temperature. J. Clim. 20, 5473–5496 (2007).
Chao, Y., Li, Z., Farrara, J. D. & Hung, P. Blending sea surface temperatures from multiple satellites and in situ observations for coastal oceans. J. Atmos. Ocean. Technol. 26, 1415–1426 (2009).
Zhang, Z., Falter, J., Lowe, R. & Ivey, G. The combined influence of hydrodynamic forcing and calcification on the spatial distribution of alkalinity in a coral reef system. J. Geophys. Res. Oceans 117, C04034 (2012).
Vengosh, A., Kolodny, Y., Starinsky, A., Chivas, A. R. & McCulloch, M. T. Coprecipitation and isotopic fractionation of boron in modern biogenic carbonates. Geochim. Cosmochim. Acta 55, 2901–2910 (1991).
Mavromatis, V., Montouillout, V., Noireaux, J., Gaillardet, J. & Schott, J. Characterization of boron incorporation and speciation in calcite and aragonite from co-precipitation experiments under controlled pH, temperature and precipitation rate. Geochim. Cosmochim. Acta 150, 299–313 (2015).
Hemming, N. G. & Hanson, G. N. Boron isotopic composition and concentration in modern marine carbonates. Geochim. Cosmochim. Acta 56, 537–543 (1992).
Zeebe, R. & Wolf-Gladow, D. A. in Elsevier Oceanography Series, vol. 65 (Elsevier, 2001).
Foster, G. L., Pogge von Strandmann, P. A. E. & Rae, J. W. B. Boron and magnesium isotopic composition of seawater. Geochem. Geophys. Geosyst. 11, Q08015 (2010).
Klochko, K., Kaufman, A. J., Yoa, W., Byrne, R. H. & Tossell, J. A. Experimental measurement of boron isotope fractionation in seawater. Earth Planet. Sci. Lett. 248, 261–270 (2006).
Dickson, A. G. Thermodynamics of the dissociation of boric acid in synthetic seawater from 273.15 to 318.15 K. Deep Sea Res. A 37, 755–766 (1990).
Sen, S., Stebbins, J. F., Hemming, N. G. & Ghosh, B. Coordination environments of B impurities in calcite and aragonite polymorphs: a 11B MAS NMR study. Am. Mineral. 79, 819–825 (1994).
Lewis, E. & Wallace, D. Program Developed for CO 2 System Calculations (Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U.S. Department of Energy, 1998).
Mehrbach, C., Culberson, C. H., Hawley, J. E. & Pytkowicz, R. N. Measurement of the apparent dissociation constants of carbonic acid in seawater at atmospheric pressure. Limnol. Oceanogr. 18, 897–907 (1973).
Dickson, A. G. & Millero, F. J. A comparison of the equilibrium constants for the dissociation of carbonic acid in seawater media. Deep Sea Res. 34, 1733–1743 (1987).
Dickson, A. G. Standard potential of the reaction: AgCl(s)+1/2H2(g)=Ag(s)+HCl(aq) and the standard acidity constant of the ion HSO4− in synthetic seawater from 273.15 to 318.15 K. J. Chem. Thermodyn. 22, 113–127 (1990).
Mucci, A. The solubility of calcite and aragonite in seawater at various salinities, temperatures, and one atmospheric total pressure. Am. J. Sci. 283, 781–799 (1985).
Acknowledgements
This research was supported by funding provided from an ARC Laureate Fellowship (LF120100049) awarded to Professor Malcolm McCulloch and the ARC Centre of Excellence for Coral Reef Studies (CE140100020). Measurements of the δ11B isotopic and B/Ca elemental ratios were conducted at the University of Western Australia’s Advanced Geochemical Facility for Indian Ocean Research (AGFIOR), and we thank Anne-Marin Comeau and Dr Kai Rankenburg for their technical assistance.
Author information
Authors and Affiliations
Contributions
M.T.M. wrote the draft of the manuscript and all authors (M.T.M., J.P.D., J.F., M.H. and J.A.T.) participated in collecting the geochemical data, analysing the results and shaping the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Data 1
Seawater (blue) and coral calcifying fluid parameters (orange). DIC = Dissolved Inorganic Carbon. pH*cf and G* are expected calcifying fluid (cf) pH and calcification rates (G) from fixed condition experimental calibrations (pH*cf = 0.32pHsw + 5.2). See manuscript for more details. (XLSX 66 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
McCulloch, M., D’Olivo, J., Falter, J. et al. Coral calcification in a changing World and the interactive dynamics of pH and DIC upregulation. Nat Commun 8, 15686 (2017). https://doi.org/10.1038/ncomms15686
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms15686
This article is cited by
-
Differences in carbonate chemistry up-regulation of long-lived reef-building corals
Scientific Reports (2023)
-
Stylasterid corals build aragonite skeletons in undersaturated water despite low pH at the site of calcification
Scientific Reports (2022)
-
Coral calcification mechanisms in a warming ocean and the interactive effects of temperature and light
Communications Earth & Environment (2022)
-
Recent ocean acidification trends from boron isotope (δ11B) records of coral: Role of oceanographic processes and anthropogenic CO2 forcing
Journal of Earth System Science (2022)
-
The indirect effects of ocean acidification on corals and coral communities
Coral Reefs (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
