
Abstract
Exploitation of plant lignocellulosic biomass is hampered by our ignorance of the molecular basis for its properties such as strength and digestibility. Xylan, the most prevalent non-cellulosic polysaccharide, binds to cellulose microfibrils. The nature of this interaction remains unclear, despite its importance. Here we show that the majority of xylan, which forms a threefold helical screw in solution, flattens into a twofold helical screw ribbon to bind intimately to cellulose microfibrils in the cell wall. 13C solid-state magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy, supported by in silico predictions of chemical shifts, shows both two- and threefold screw xylan conformations are present in fresh Arabidopsis stems. The twofold screw xylan is spatially close to cellulose, and has similar rigidity to the cellulose microfibrils, but reverts to the threefold screw conformation in the cellulose-deficient irx3 mutant. The discovery that induced polysaccharide conformation underlies cell wall assembly provides new principles to understand biomass properties.
Similar content being viewed by others


Introduction
Plant secondary cell walls are strong composites of polysaccharides, lignin and proteins that are crucial for plant structure. They provide the most abundant renewable materials on Earth. This lignocellulosic material is therefore the focus of biofuel development1, and wood is widely used for many industries such as building construction, paper and packaging. Polysaccharides constitute over 70% of dry cell wall lignocellulosic biomass and can be categorized as cellulose, hemicelluloses and pectins in decreasing order of prevalence (Supplementary Fig. 1)2,3. Properties such as biomass recalcitrance and timber strength arise from secondary cell wall molecular architecture: the molecular arrangements and conformations of the polysaccharides and other components within the secondary cell wall. Nevertheless, cell wall molecular architecture is very poorly understood.
The semi-crystalline cellulose microfibrils provide the best understood example of molecular architecture in lignocellulose. Its component β-(1→4)-D-glucan chains fold in a twofold helical screw conformation (one 360° twist per 2 glycosidic bonds). The pattern of hydrogen bonding interactions is well understood from X-ray diffraction and magic-angle spinning (MAS) solid-state nuclear magnetic resonance (NMR) studies4,5,6. On the other hand, the intermolecular interactions that occur between cellulose fibrils and hemicelluloses have been a matter of debate for several decades2,7. Xyloglucan was thought to crosslink microfibrils in primary cell walls. Multidimensional MAS NMR analyses of primary cell walls, pioneered in recent years by Hong and colleagues, have shown that, in contrast, only a minor portion of xyloglucan interacts with cellulose8,9,10,11,12,13,14. In secondary cell walls, xylan (Supplementary Fig. 1) and glucomannan are known to bind to cellulose microfibrils, but there is little evidence for the mechanism of this interaction. As the main hemicellulose of eudicot secondary cell walls, the interaction of xylan with cellulose is expected to have the most profound effect on cell wall characteristics and therefore properties such as biomass recalcitrance and timber strength. Indeed, Arabidopsis plants with reduced xylan quantity show weakened walls and the plants are unable to develop a vascular system15,16. Molecular dynamics simulations suggest multiple layers of xylan could envelop microfibrils17, and reconstitution studies have revealed that xylan is able to crosslink microfibrils18, yet no clear model for the interaction has emerged. On the basis of a conserved pattern of xylan substitution strictly on alternate xylosyl residues, it has recently been proposed that xylan might be able to hydrogen bond to the hydrophilic surfaces of cellulose through folding as a twofold helical screw (Supplementary Fig. 2)19,20,21,22. This model is controversial because xylan in solution forms a threefold helical screw19,23 (one 360° twist per three glycosidic bonds). Despite the conservation of a periodic structure of xylan substitutions throughout seed plants, there is no direct experimental evidence for this model. MAS NMR has the potential to reveal polysaccharide conformations and interactions11,24,25,26,27. We have therefore recently developed its use to study Arabidopsis secondary cell wall28, so that mutants can be studied.
By combining Arabidopsis molecular genetics and MAS NMR approaches, here we show that xylan interacts intimately with cellulose by adopting a twofold helical screw conformation. The change of xylan fold is induced by, and dependent upon, cellulose fibrils. This provides the first direct experimental evidence for the atomic-scale arrangement of two distinct polymers within plant cell wall biomass, and reveals new principles underlying the remarkable specificity of polysaccharide interactions. Our work provides an understanding of the molecular architectural basis for the biological and mechanical function of cell wall components in plant biomass.
Results
Xylan adopts two distinct conformations in the cell wall
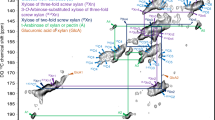
To analyse the structure of xylan in never-dried cell walls by 13C MAS NMR, a chamber to grow uniformly labelled Arabidopsis plants in 13CO2 was designed and constructed (Supplementary Fig. 3). Fresh and unprocessed stems were directly analysed, ensuring that the native arrangement of the cell wall polysaccharides was preserved. Figure 1a shows a two-dimensional (2D) refocused INADEQUATE25,29,30 MAS NMR spectrum of wild-type Arabidopsis stems using cross polarization (CP) excitation, which emphasizes the more rigid components. In this experiment directly bonded nuclei appear at the same double-quantum (DQ) chemical shift that corresponds to the sum of the two single-quantum (SQ) chemical shifts. The spectrum shows broad line widths of ∼2 p.p.m. indicating the relative rigidity of the polysaccharides and a range of different environments. As expected, the major peaks in the spectrum are from glucose in the two main cellulose environments (C1: often assigned to crystalline and/or internal glucan chains; and C2: often assigned to amorphous and/or surface glucan chains)8,9,10,11,12,28. Surprisingly, two distinct xylan carbon 4 (Xn4) peaks are clearly visible in the spectrum (82.2 and 77.4 p.p.m.), each coupled to Xn5 (64.3 and 63.9 p.p.m.). The existence of the two discrete Xn4 shifts indicates the presence of two distinct conformations of xylan in the fresh hydrated cell walls. This is in contrast to the range of shifts observed for Xn4 in dried Arabidopsis stems28, suggesting that drying had a substantial effect on native xylan conformations.

(a) The carbohydrate region of a refocussed CP-INADEQUATE 13C NMR spectrum of never-dried wild-type Arabidopsis stems is shown. Red and green labelling illustrates the two distinct xylan conformations, shown clearly here by the distinct xylan carbon 4 (Xn4) and Xn5 shifts. Xn1, Xn2 and Xn3 also differ between conformations but not all differences are readily distinguished in this CP spectrum (see Fig. 4 and Table 1 for full assignments). The spectrum was recorded at a 13C Larmor frequency of 125.8 MHz and a MAS frequency of 10 kHz using a total spin-echo duration of 1.2 ms. (b) In silico DFT-optimized xylan structures from which 13C chemical shifts in Table 1 were calculated.
Xylan in solution forms a threefold helical screw19,23. The 77.4 p.p.m. Xn4 and 63.9 p.p.m. Xn5 shifts are very similar to the solution state xylan Xn4 shift (Table 1), suggesting that they arise from threefold screw xylan in the cell wall, so we named them Xn43f and Xn53f, respectively. It has been proposed that some xylan may fold as twofold screw in the cell wall19,20,21,22. A change in torsion angle around the glycosidic bond (which yields different helical conformations) can alter the chemical shifts of polysaccharides, and in (1→4)-linked glycans changes in torsion angle has been shown to affect carbon 1 and carbon 4 resonances particularly27. The 82.2 p.p.m. Xn4 shift seen in the fresh cell walls might therefore arise from a change in conformation of the xylan to a twofold screw. To estimate the shift differences between the twofold screw and threefold screw xylan conformations, we performed in silico13C chemical shift prediction using gauge including projector augmented waves density functional theory (DFT)31,32 (Fig. 1b). As expected, the two carbons involved in the glycosidic bond–Xn1 and Xn4–were predicted to alter the most after changing conformation (Table 1 and Supplementary Tables 1 and 2). Xn1 and Xn4 are predicted to be substantially shifted to higher p.p.m. in a twofold screw compared with a threefold screw conformation. Dominant cellulose signals mask the twofold Xn1 signal (Xn12f) in the CP-refocused INADEQUATE spectrum (Fig. 1; Xn1 assigned below). However, the presence of the Xn4 signal at the higher (82.2 p.p.m., Xn42f) 13C chemical shift, covalently linked to an Xn5 signal (Xn52f), indicates that xylan forms a twofold screw in the cell wall.
Cellulose is required for xylan twofold screw conformation
Xylan might be induced to adopt the twofold screw conformation upon binding to cellulose in the cell wall19,20,21,22. Indeed, 82–84 p.p.m. peaks have previously been proposed to arise from cellulose-aggregated xylan33,34,35,36. Therefore, we next investigated whether the twofold screw xylan is dependent on cellulose in the cell wall. The irx3 Arabidopsis mutant is deficient in secondary cell wall cellulose synthesis and is consequently dwarfed, but the plant continues to synthesize other cell wall components including xylan37,38. Because it has much reduced secondary cell wall cellulose, it provides a useful tool to study the importance of cellulose for xylan conformation. One-dimensional MAS NMR of irx3 stems confirmed depletion in this mutant of cellulose and the relative increase in other cell wall polysaccharides, including xyloglucan (Xg) and pectin (GalA) (Supplementary Fig. 4). Interestingly, a CP-refocused INADEQUATE spectrum showed a profound difference in the xylan conformation in irx3 compared with wild type. While twofold screw xylan (Xn42f peak) was by far the more prevalent of the two conformations in wild type (Figs 1 and 2), in irx3 it was barely detectable (Fig. 2). In contrast, almost all irx3 xylan was found in the threefold conformation. To study the more mobile cell wall components in irx3 and wild-type stems, direct polarization (DP)-INADEQUATE spectra with a short recycle delay were acquired. The abundance of the Xn43f peak showed that relatively mobile threefold screw xylan is far more prevalent in irx3 than in the wild type (Fig. 3 and Supplementary Fig. 4). Indeed, this mobile threefold screw xylan was not detected in wild-type plants. Neither the twofold screw xylan nor cellulose was seen in the DP-INADEQUATE spectra of wild-type plants, indicating that the twofold screw xylan, like cellulose, is relatively immobile. The absence of the twofold screw xylan in irx3 shows that the change in xylan conformation from threefold to twofold screw is cellulose-dependent.

An overlay is shown of carbohydrate regions of refocussed CP-INADEQUATE 13C NMR spectra of wild-type and irx3. Red and green labelling illustrates the two distinct xylan conformations shown clearly here by the distinct Xn4 and Xn5 shifts. The spectra were recorded as in Fig. 1, using a total spin-echo duration of 2.2 ms.

An overlay is shown of 13C refocussed DP-INADEQUATE 13C NMR spectra of wild type and irx3 never-dried Arabidopsis stems using a 1.9 s recycle time, which emphasize relatively mobile species. The carbohydrate region of the spectrum is expanded to focus on the Xn4→Xn5 connection. This shows that threefold xylan is abundant in irx3, and the twofold xylan is relatively immobile and so is not detected in wild type or irx3 mutant cell walls. Xylose of threefold xylan is highlighted in green. Dotted red squares indicate the absence of the twofold screw Xn4→Xn5 pair. The change in some arabinose and pectin structures in the plants may be due to altered growth of the dwarfed irx3 plant. The spectrum of the full carbohydrate region is shown in Supplementary Figure 5, and was recorded with a spin-echo duration of 2.2 ms at a 13C Larmor frequency of 213.8 MHz and a MAS frequency of 12 kHz.
The presence of abundant threefold screw xylan in irx3 cell walls gave us the opportunity to investigate further the Xn12f and Xn13f 13C chemical shifts, given the DFT prediction of substantial change in Xn1 (Table 1). Using the knowledge that xylan is highly acetylated, the shifts of carbon atoms close to acetate moieties in the cell wall were investigated using proton-driven spin diffusion (PDSD) experiments, which measure through-space proximities of different moieties39, with longer mixing times probing longer distances. Figure 4 shows a comparison of long mixing time PDSD spectra of wild-type plants (a CP spectrum where mainly twofold xylan is visible), with irx3 (a DP-PDSD spectrum where only threefold xylan is visible). Acetate methyl groups (21.6 p.p.m.) are spatially close to acetate carbonyl groups (173.6 p.p.m.) and also to Xn1–Xn5. Interestingly, the Xn13f shift in irx3 is 102.6 p.p.m. as seen in DP INADEQUATE and in solution xylan, but in wild-type Xn12f is significantly shifted to the higher 13C chemical shift of 105.2 p.p.m.. Because a clear increase in Xn12f shift from threefold to twofold was predicted by the calculations (Table 1), this observation further supports the assignment of the conformation which predominates in wild type as twofold screw, and the conformation which predominates in irx3 as threefold. Notably, the Xn12f shift of 105.2 p.p.m. is very similar to cellulose C1 at 105.1 p.p.m., which also exists in a twofold screw.

An overlay is shown of 13C PDSD NMR spectra of wild type (CP-PDSD 1 s mixing time) and irx3 (DP-PDSD, 1.5 s mixing time) never-dried Arabidopsis stems. Wild-type and irx3 show two distinct xylan conformations: in wild-type plants Xn12f resonates at 105.2 p.p.m., whereas in irx3 Xn13f is at 102.6 p.p.m. These long mixing time spectra were recorded at a 13C Larmor frequency of 213.8 MHz and a MAS frequency of 12 kHz.
Xylan with a twofold screw conformation is bound to cellulose
To determine whether the twofold screw xylan is cellulose-bound, spatial proximities of molecules in the cell wall were investigated using a relaxation compensated z-filtered version of the CP-PDSD experiment40 (Fig. 5). In the experiment with a short 50 ms mixing time, cross peaks indicative of short-distance intramolecular spatial relationships were observed, for example, within cellulose domain 1 (C41-C61; 89.0 p.p.m.-65.3 p.p.m.), cellulose domain 2 (C42-C62; 84.1 p.p.m. to 62.6 p.p.m.) and twofold screw xylan (Xn42f-Xn52f; 82.2 p.p.m. to 64.2 p.p.m.; Supplementary Table 1). In the experiment with a longer, 1 s, mixing time cross peaks indicative of both short and longer distance intra- and intermolecular spatial relationships were observed. Interestingly, cross peaks were seen between xylan and cellulose, indicating their close spatial proximity. However, xylan cross peaks to cellulose domain 1 (for example, Xn42f→C61; Fig. 5c,d), which is often assigned to internal cellulose glucan chains, were greater than those to cellulose domain 2 (for example, Xn42f→C62 Fig. 5c), which is often assigned to surface cellulose glucans. This suggests that xylan coating of cellulose surface chains causes a change in glucan NMR shift, perhaps because these glucans adopt an interior-like conformation, akin to a recent proposal for cellulose: hemicellulose interactions in primary cell walls13. The similar signal strengths of the intermolecular Xn42f→C61 and the intramolecular Xn42f→Xn52f peaks indicate that almost all of the twofold screw xylan is spatially close to cellulose. In contrast at this mixing time the cross peaks showing spatial proximities between cellulose domains (for example, C41→C62 and C61→C62) are not yet as strong as those showing spatial proximities within cellulose domains (for example, C41→C61). Although spin diffusion in a fully 13C labelled system is complex, the presence of the relatively large Xn42f→C61 cross peak means that we can place an upper limit on the distance between the twofold screw xylan and cellulose: twofold screw xylan can be no farther than the width of a microfibril from the cellulose surface. Hence, the PDSD experiments indicate that the twofold screw xylan is bound to cellulose.

Carbohydrate regions are shown of z-filtered CP-PDSD39 13C NMR spectra of never-dried wild-type Arabidopsis stems using a total z period of 1.005 s. (a) 50 ms mixing time spectrum. (b) 1 s mixing time spectrum. (c) Zoom of 1 s mixing time spectrum showing cellulose→xylan connections. (d) Zoom of 1 s mixing time spectrum showing xylan→cellulose connections. (e) One-dimensional slices from 50 ms (blue) and 1 s (red) mixing time spectra. The spectra were recorded as in Fig. 4.
As a further means of probing xylan: cellulose interactions we analysed the molecular mobility of these polysaccharides by measuring the 13C T1 relaxation time (Supplementary Table 3), which is sensitive to motion on the nanosecond timescale, and the dipolar order parameter SCH (0≤SCH≤1) which is sensitive to motion on the microsecond timescale. Both DP and CP DIPSHIFT41 experiments were used to measure the order parameter as there is more than one contribution to the signal for many chemical shifts. The DP DIPSHIFT with 20 s recycle delay was quantitative with all of the species contributing, whereas the CP DIPSHIFT preferentially measured the immobile components such as cellulose and any cellulose-bound xylan. Figure 6 shows a comparison of the CP order parameters representative of cellulose, twofold xylan and threefold xylan, all obtained from the dephasing curves shown in Supplementary Fig. 6, for both wild-type and irx3 samples. Cellulose-bound xylan would be expected to exhibit less motion (demonstrated by a larger order parameter) than unbound xylan. The order parameter of twofold xylan (specifically measuring Xn42f in wild-type stems) is comparable to that of both cellulose domains (specifically measuring C41 and C42 in wild-type stems), indicating relatively little motion. In contrast, for threefold xylan (specifically measuring Xn13f and Xn43f in irx3 stems) the order parameter is significantly reduced, indicating more motion. Consistent with this, the T1 of Xn42f (82.5 p.p.m.) in wild type is very similar to cellulose values whereas Xn13f in irx3 has shorter T1 (see Supplementary Table 3). Therefore, twofold screw xylan is far less mobile than threefold screw xylan, and has a similar rigidity to cellulose.

CP-DIPSHIFT MAS NMR analysis of the order parameter, SCH, in wild-type and irx3 stems is shown; this reports on the molecular mobilities of cell wall moieties. The labelling refers to the presumed major contributing species. Refer to Supplementary Fig. 4 for chemical shift assignments of the One-dimensional spectra. See Supplementary Fig. 6 for data fits. The error bars were determined by simulation of the DIPSHIFT curves shown in Supplementary Fig. 6b with varying parameters. The error bar represents the maximum change in parameter before there is a clear deviation of simulated from experimental values. The order parameters for all major peaks are shown in Supplementary Fig. 7. Experiments were carried out at a 13C Larmor frequency of 125.8 MHz and a MAS frequency of 7.813 kHz.
The CP-INADEQUATE and DP-INADEQUATE experiments together indicated that the large majority of xylan in the secondary cell walls of wild-type plants folds as twofold screw, with only a minor proportion of xylan detected in a relatively immobile threefold screw conformation. The PDSD and DIPSHIFT experiments, together with the requirement for cellulose revealed by irx3, indicated that essentially all the twofold screw xylan is bound to cellulose. These results are consistent with the finding that the majority of xylan has substitution patterning that is compatible with binding to the hydrophilic surfaces of cellulose19,20,21. However, we do not yet know the relative amounts of the xylan bound to the hydrophilic vs hydrophobic surfaces of the fibrils. Moreover, the proportion of hydrophilic and hydrophobic surfaces on the cellulose microfibril structure is not yet known. In the secondary cell walls of eudicots, the ratio between quantity of xylan and cellulose is approximately 2:3 (ref. 42). Our results therefore suggest that there is likely sufficient xylan to coat the hydrophilic surfaces of cellulose microfibrils. Nevertheless, further work is necessary to determine the extent of xylan binding to the different fibril surfaces.
Discussion
The molecular arrangement of plant cell wall components has been an unresolved question for decades. We have now shown that xylan is induced to assemble on cellulose fibrils as a twofold helical screw in secondary cell walls. This is the first direct evidence for the atomic-scale model of the interactions between cellulose and xylan in plant cell walls (Supplementary Fig. 2)19,20. In this model, xylan hydrogen bonds with cellulose microfibril hydrophilic surfaces by forming a flattened twofold helical screw. This effectively extends the semi-crystalline cellulose microfibril size and alters its properties. Coating of the hydrophilic faces of cellulose microfibrils with the acetylated and glucuronosylated form of xylan found in dicot cell walls may lead the otherwise hydrophilic surfaces to be relatively hydrophobic (because of acetate groups) and acidic (because of glucuronic acid groups). This may be of profound importance to cell wall architecture, for example influencing interactions between the cellulose-xylan fibrils, and altering binding to the other components of the secondary cell wall, especially lignin. Xylan folded onto cellulose surfaces is likely to be resistant to the action of microbial hydrolases which cleave threefold screw xylan. It remains unknown whether some xylan may also bind to the hydrophobic surface of fibrils. The evenly spaced glucuronosyl and acetyl substitutions of xylan19,20,21,22 are important for allowing xylan to bind to the hydrophilic surfaces of cellulose. Because xylan from gymnosperms to eudicots has retained this even pattern of substitution22, we believe the assembly of xylan onto cellulose revealed in this work is likely to be conserved across land plants. Through discovery of the specific interaction of these two polysaccharides, this work therefore reveals a fundamental principle in assembly of plant cell walls which likely extends to other polysaccharide interactions and the extracellular matrix of many organisms.
A description of the nature of the interactions between cell wall components is crucial for many uses of plants as a renewable resource. It helps us understand the basis of plant recalcitrance to digestibility and deconstruction, which is of importance in dietary fibre, animal fodder and in biofuel production. These findings will also be important in improving processes using materials made from plants, such as the paper industry and building construction. Our studies of cell wall molecular architecture therefore provide a new twist for exploitation of plant materials.
Methods
Growth chamber and plant preparation
irx3-7 seeds were kindly donated by Simon Turner (University of Manchester). Seeds were surface sterilized and sown on 0.8% (w/v) agar, 0.5 9 Murashige and Skoog salts including vitamins (Sigma, http://www.sigma.com) and sucrose (1% w/v). Following stratification for 48 h at 4 °C in the dark, plates were transferred to a growth room (20 °C, 100 μmol m−2 s−1, 24 h light, 60% humidity). Optimaxx fibre rockwool (Cultilene, Netherlands) was cut into slabs and laid ∼8 cm deep in a growth tray. Hydroponics solution (2 mM MgSO4, 2 mM CaNO3, 50 μM FeEDTA, 5 mM KNO3, 2.5 mM K2HPO4/KH2PO4 pH 5.5, 70 μM H3BO4, 14 μM MnCl2, 0.5 μM CuSO4, 1 μM ZnSO4, 0.2 μM NaMoO4, 10 μM NaCl and 0.1 μM CoCl2) was poured into the tray until half-way up. The rockwool was then covered with foil. Holes were pierced into the foil and rockwool and seedling were placed in, ensuring that roots made direct contact with rockwool. A growth chamber was constructed following Chen et al.43 illustrated in Supplementary Fig. 3. Compressed air was scrubbed of CO2 using calcium oxide before 13CO2 was mixed back in at a concentration of 500 p.p.m. before entering the growth chamber. Plants were grown in a 50% humidity and 24 °C environment for 6–8 weeks. 13C enrichment was measured by analysing xyloglucan oligosaccharides yielded from xyloglucan endoglucanase digestion of stems using matrix-assisted laser desorption/ionization–time of flight mass spectrometry. Typical enrichments were 90–95%. Roughly 35 mg of the bottom third parts of five to ten stems were chopped and packed into a 3.2 mm Magic-Angle Spinning NMR rotor. For wild-type, five biological replicates (of at least five plants) were grown, each of which was analysed at least once; for irx3, two biological replicates (of at least five plants) were grown, each of which was analysed at least once. On the basis of the relative content of cellulose, pectin, xyloglucan and xylan observed in the wild-type and irx3 samples by NMR, we estimate the stem material contained a substantial but minority proportion of primary cell walls.
NMR chemical shift predictions
The NMR shift calculations were carried out using the CASTEP code44 with the PBE45 exchange correlation functional. The 10 molecule (two- and threefold) MD xylan structures generated earlier19 were first geometry optimized using a cutoff energy of 800 eV with a k point grid of 2 × 2 × 1 and a fixed unit cell of 10 × 10 × 60 Å. The NMR shielding was calculated for the resulting structures using the gauge including projector augmented waves31,32 method. The reference used to convert shielding to shift, 167.7 p.p.m., was determined by assuming that the threefold structure is very similar to xylan in solution (see Supplementary Table 2). Its value does not affect the shift differences given in Table 1.
Solid-state NMR
Solid-state MAS NMR experiments were performed on Bruker (Karlsruhe, Germany) 850 and 500 MHz Advance III solid-state NMR spectrometers, operating at 1H and 13C Larmor frequencies of 850.2 and 213.8 MHz and 500.1 and 125.8 MHz, respectively, using 3.2 mm double-resonance MAS probes. Experiments were conducted at room temperature at MAS frequencies of 12–14 kHz at the higher field and 10 kHz on the 500 MHz spectrometer unless otherwise stated. The 13C chemical shift was determined using the carbonyl peak at 177.8 p.p.m. of L-alanine as an external reference with respect to tetramethylsilane (TMS); 90° pulse lengths were typically 3.5 μs (1H) and 4.2 μs (13C). Both 1H−13C cross-polarization (CP) with ramped (70–100%) 1H rf amplitude46 and a contact time of 1 ms and direct polarization (DP) were used to obtain the initial transverse magnetization. While CP emphasizes the more rigid material, a short, 1.9 s, recycle delay DP experiment was used to preferentially detect the mobile components and a 20 s delay was used for quantitative experiments. SPINAL-64 decoupling47 was applied during acquisition at a 1H nutation frequency of 70–80 kHz. Two-dimensional double-quantum (DQ) correlation spectra were recorded using the refocused INADEQUATE pulse sequence which relies upon the use of isotropic, scalar J coupling to obtain through-bond information regarding directly coupled nuclei25,29,30. The carbon 90° and 180° pulse lengths were 4 and 8 μs, respectively with 2τ spin-echo evolution times for a (τ–π–τ) spin-echo of 1.2 to 4.4 ms and the SPINAL-64 1H decoupling47 was applied during both the evolution and signal acquisition periods. The acquisition time in the indirect dimension (t1) was 4.5 -5.0 ms with a recycle delay of 1.5 and 1.9 s for CP and DP experiments, respectively. Intermolecular contacts were probed using 2D 13C−13C 1H driven spin diffusion (PDSD) experiments39,40 with mixing times of 50 ms to 1.5 s. For both refocused INADEQUATE and PDSD experiments, the spectra were obtained by Fourier transformation into 2 K (F2) × 1 K (F1) points with exponential line broadening of 80 Hz (CP) and 40 Hz (DP) in F2 and squared sine bell processing in F1. The 13C spin lattice relaxation time, T1, was measured at 125.8 MHz using saturation recovery following a comb of 30 pulses and echo acquisition. The dipolar order parameter, SCH, was determined using the 2D 13C−1H dipolar chemical shift (DIPSHIFT) experiment41 whilst spinning at 7.813 kHz on the 500 MHz spectrometer. The 1H homonuclear coupling was suppressed using the frequency switched Lee-Goldberg sequence48 with a 1H nutation frequency of 80 kHz. Measurements were taken with CP and with DP excitation with recycle delays of 2 and 20 s, respectively. All spectra obtained were processed and analysed using Bruker Topspin version 3.2.
DIPSHIFT calculations
DIPSHIFT experiments were simulated using a home written program in Origin.c. The DIPSHIFT curves were calculated as a simple evolution of the 13C magnetization under C−H dipolar coupling and MAS. The effective C−H coupling for CH2 units was obtained using the expressions reported by Terao et al.49 This provides a reliable measurement of the effective C−H coupling in the fast and rigid motion limits, as confirmed by comparison with full spin dynamics simulations50.
Data availability
The solid-state NMR data and scripts for DIPSHIFT simulations are available at http://dx.doi.org/10.17863/CAM.5896. The authors declare that all other relevant data supporting the findings of this study are available within the article and its Supplementary Information files or on request from the corresponding authors.
Additional information
How to cite this article: Simmons, T. J. et al. Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat. Commun. 7, 13902 doi: 10.1038/ncomms13902 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Perlack, R. D. & Stokes, B. J., Leads. U.S. Billion-Ton Update: Biomass Supply for a Bioenergy and Bioproducts Industry. Technical Report ORNL/TM-2011/224, 227. (U.S. Department of Energy, Oak Ridge National Laboratory, Oak Ridge, TN, 2011).
Carpita, N. & McCann, M. in Biochemistry and Molecular Biology of Plants, Vol. 61 (eds Buchanan, B. B., Wilhelm, G., and Jones, R. L.) 52–108, (American Society of Plant Physiologists, Rockville, IL, 2000).
Scheller, H. V. & Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 61, 263–289 (2010).
Atalla, R. H. & Vanderhart, D. L. Native cellulose - a composite of 2 distinct crystalline forms. Science 223, 283–285 (1984).
Nishiyama, Y., Langan, P. & Chanzy, H. Crystal structure and hydrogen-bonding system in cellulose 1β from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 124, 9074–9082 (2002).
Nishiyama, Y., Sugiyama, J., Chanzy, H. & Langan, P. Crystal structure and hydrogen bonding system in cellulose 1α, from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 125, 14300–14306 (2003).
Cosgrove, D. J. & Jarvis, M. C. Comparative structure and biomechanics of plant primary and secondary cell walls. Front. Plant Sci. 3, 1–6 (2012).
Dick-Perez, M. et al. Structure and Interactions of plant cell-wall polysaccharides by two- and three-dimensional magic-angle-spinning solid-state NMR. Biochemistry 50, 989–1000 (2011).
Dick-Perez, M., Wang, T., Salazar, A., Zabotina, O. A. & Hong, M. Multidimensional solid-state NMR studies of the structure and dynamics of pectic polysaccharides in uniformly C-13-labeled Arabidopsis primary cell walls. Magn. Reson. Chem. 50, 539–550 (2012).
Wang, T., Zabotina, O. & Hong, M. Pectin-cellulose interactions in the arabidopsis primary cell wall from two-dimensional magic-angle-spinning solid-state nuclear magnetic resonance. Biochemistry 51, 9846–9856 (2012).
Wang, T. et al. Sensitivity-enhanced solid-state NMR detection of expansin's target in plant cell walls. Proc. Natl Acad. Sci. USA 110, 16444–16449 (2013).
Wang, T., Salazar, A., Zabotina, O. A. & Hong, M. Structure and dynamics of brachypodium primary cell wall polysaccharides from two-dimensional 13C solid-state nuclear magnetic resonance spectroscopy. Biochemistry 53, 2840–2854 (2014).
Wang, T., Yang, H., Kubicki, J. D. & Hong, M. Cellulose structural polymorphism in plant primary cell walls investigated by high-field 2D Solid-state NMR spectroscopy and density functional theory calculations. Biomacromolecules 17, 2210–2222 (2016).
Bootten, T. J., Harris, P. J., Melton, L. D. & Newman, R. H. Solid-state 13C-NMR spectroscopy shows that the xyloglucans in the primary cell walls of mung bean (Vigna radiata L.) occur in different domains: a new model for xyloglucan-cellulose interactions in the cell wall. J. Exp. Bot. 55, 571–583 (2004).
Wu, A. M. et al. The Arabidopsis IRX10 and IRX10-LIKE glycosyltransferases are critical for glucuronoxylan biosynthesis during secondary cell wall formation. Plant J. 57, 718–731 (2009).
Brown, D. M. et al. Comparison of five xylan synthesis mutants reveals new insight into the mechanisms of xylan synthesis. Plant J. 52, 1154–1168 (2007).
Li, L., Perre, P., Frank, X. & Mazeau, K. A coarse-grain force-field for xylan and its interaction with cellulose. Carbohyd. Polym. 127, 438–450 (2015).
Mikkelsen, D., Flanagan, B. M., Wilson, S. M., Bacic, A. & Gidley, M. J. Interactions of arabinoxylan and (1,3)(1,4)-β-glucan with cellulose networks. Biomacromolecules 16, 1232–1239 (2015).
Busse-Wicher, M. et al. The pattern of xylan acetylation suggests xylan may interact with cellulose microfibrils as a twofold helical screw in the secondary plant cell wall of Arabidopsis thaliana. Plant J. 79, 492–506 (2014).
Busse-Wicher, M., Grantham, N. J., Lyczakowski, J. J., Nikolovski, N. & Dupree, P. Xylan decoration patterns and the plant secondary cell wall molecular architecture. Biochem. Soc. Trans. 44, 74–78 (2016).
Bromley, J. R. et al. GUX1 and GUX2 glucuronyltransferases decorate distinct domains of glucuronoxylan with different substitution patterns. Plant J. 74, 423–434 (2013).
Busse-Wicher, M. et al. Evolution of xylan substitution patterns in gymnosperms and angiosperms: implications for xylan interaction with cellulose. Plant Physiol. 171, 2418–2431 (2016).
Nieduszynski, I. & Marchessault, R. H. Structure of β-D-(1→4')Xylan Hydrate. Nature 232, 1335–1344 (1971).
Bardet, M., Emsley, L. & Vincendon, M. Two-dimensional spin-exchange solid-state NMR studies of 13C-enriched wood. Solid State Nucl. Magn. Reson. 8, 25–32 (1997).
Lesage, A., Bardet, M. & Emsley, L. Through-bond carbon-carbon connectivities in disordered solids by NMR. J. Am. Chem. Soc. 121, 10987–10993 (1999).
Komatsu, T. & Kikuchi, J. Selective signal detection in solid-state NMR Using rotor-synchronized dipolar dephasing for the analysis of hemicellulose in lignocellulosic biomass. J. Phys. Chem. Lett. 4, 2279–2283 (2013).
Jarvis, M. C. & Apperley, D. C. Chain conformation in concentrated pectic gels - evidence from 13C NMR. Carbohyd. Res. 275, 131–145 (1995).
Dupree, R. et al. Probing the molecular architecture of Arabidopsis thaliana secondary cell walls using two- and three-dimensional 13C solid state nuclear magnetic resonance spectroscopy. Biochemistry 54, 2335–2345 (2015).
Lesage, A., Auger, C., Caldarelli, S. & Emsley, L. Determination of through-bond carbon-carbon connectivities in solid-state NMR using the INADEQUATE experiment. J. Am. Chem. Soc. 119, 7867–7868 (1997).
Fayon, F. et al. Through-space contributions to two-dimensional double-quantum J correlation NMR spectra of magic-angle-spinning solids. J. Chem. Phys. 122, 14931 (2005).
Pickard, C. J. & Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 63, 245101 (2001).
Yates, J. R., Pickard, C. J. & Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 76, 024401 (2007).
Wickholm, K., Larsson, P. T. & Iversen, T. Assignment of non-crystalline forms in cellulose I by CP/MAS 13C NMR spectroscopy. Carbohyd. Res. 312, 123–129 (1998).
Larsson, P. T., Hult, E. L., Wickholm, K., Pettersson, E. & Iversen, T. CP/MAS C-13-NMR spectroscopy applied to structure and interaction studies on cellulose I. Solid State Nucl. Magn. Reson. 15, 31–40 (1999).
Teleman, A., Larsson, P. T. & Iversen, T. On the accessibility and structure of xylan in birch kraft pulp. Cellulose 8, 209–215 (2001).
Habibi, Y., Heux, L., Mahrouz, M. & Vignon, M. R. Morphological and structural study of seed pericarp of Opuntia ficus-indica prickly pear fruits. Carbohyd. Polym. 72, 102–112 (2008).
Ha, M. A. et al. Structure of cellulose-deficient secondary cell walls from the irx3 mutant of Arabidopsis thaliana. Phytochemistry 61, 7–14 (2002).
Brown, D. M., Zeef, L. A. H., Ellis, J., Goodacre, R. & Turner, S. R. Identification of novel genes in Arabidopsis involved in secondary cell wall formation using expression profiling and reverse genetics. Plant Cell 17, 2281–2295 (2005).
Takegoshi, K., Nakamura, S. & Terao, T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637 (2001).
Wang, T., Williams, J. K., Schmidt-Rohr, K. & Hong, M. Relaxation-compensated difference spin diffusion NMR for detecting 13C-13C long-range correlations in proteins and polysaccharides. J. Biomol. NMR 61, 97–107 (2015).
Munowitz, M. G., Griffin, R. G., Bodenhausen, G. & Huang, T. H. Two-dimensional rotational spin-echo nuclear magnetic-resonance in solids - correlation of chemical-shift and dipolar interactions. J. Am. Chem. Soc. 103, 2529–2533 (1981).
Pauly, M. & Keegstra, K. Cell-wall carbohydrates and their modification as a resource for biofuels. Plant J. 54, 559–568 (2008).
Chen, W. P. et al. An automated growth enclosure for metabolic labeling of Arabidopsis thaliana with 13C-carbon dioxide - an in vivo labeling system for proteomics and metabolomics research. Proteome Sci. 9, 9 (2011).
Clark, S. J. et al. First principles methods using CASTEP. Z. Kristallogr 220, 567–570 (2005).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Metz, G., Wu, X. L. & Smith, S. O. Ramped-amplitude cross-polarization in magic-angle-spinning NMR. J. Magn. Reson. Ser. A 110, 219–227 (1994).
Fung, B. M., Khitrin, A. K. & Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 142, 97–101 (2000).
Bielecki, A., Kolbert, A. C. & Levitt, M. H. Frequency-switched pulse sequences - homonuclear decoupling and dilute spin NMR in solids. Chem. Phys. Lett. 155, 341–346 (1989).
Terao, T., Miura, H. & Saika, A. Dipolar SASS NMR-spectroscopy - separation of heteronuclear dipolar powder patterns in rotating solids. J. Chem. Phys. 85, 3816–3826 (1986).
DeAzevedo, E. R. et al. Intermediate motions as studied by solid-state separated local field NMR experiments. J. Chem. Phys. 128, 128–139 (2008).
Acknowledgements
We thank Dinu Iuga, Shazeaa Ishmael, Howard Griffiths and Moritz Meyer for their help. This work was part supported by BBSRC Grant BB/G016240/1 via The BBSRC Sustainable Bioenergy Cell Wall Sugars Programme. ODB and ERdA are grateful to CNPq for financial support for this work via Grants # 159341/2011-6 and 206278/2014-4. ACP is grateful to the Royal Society for a Newton International Fellowship. PD is supported by the Leverhulme Trust grant for the Centre for Natural Material Innovation. The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC, as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). (Contract reference PR140003 for work after 5 January 2015). DFT calculations of NMR parameters were performed at the Centre for Scientific Computing at the University of Warwick.
Author information
Authors and Affiliations
Contributions
T.J.S. grew plants. J.C.M. designed and constructed the growth chamber. R.D., O.B. and T.J.S. conducted the NMR experiments. T.J.S., J.C.M., O.B., S.P.B., E.A., R.D. and P.D. analysed the experimental data. A.C.P. performed in silico NMR chemical shift predictions. T.J.S., R.D. and P.D. wrote the manuscript. All authors contributed critical thoughts.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures, Supplementary Tables and Supplementary References. (PDF 778 kb)
Supplementary Information
Peer Review File (PDF 290 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Simmons, T., Mortimer, J., Bernardinelli, O. et al. Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat Commun 7, 13902 (2016). https://doi.org/10.1038/ncomms13902
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms13902
This article is cited by
-
Enzymatic debranching is a key determinant of the xylan-degrading activity of family AA9 lytic polysaccharide monooxygenases
Biotechnology for Biofuels and Bioproducts (2023)
-
Deciphering triterpenoid saponin biosynthesis by leveraging transcriptome response to methyl jasmonate elicitation in Saponaria vaccaria
Nature Communications (2023)
-
Callose integration into secondary cell walls modifies woody biomass ultrastructure and accessibility
Nature Plants (2023)
-
Electron tomography unravels new insights into fiber cell wall nanostructure; exploring 3D macromolecular biopolymeric nano-architecture of spruce fiber secondary walls
Scientific Reports (2023)
-
Solution-state nuclear magnetic resonance spectroscopy of crystalline cellulosic materials using a direct dissolution ionic liquid electrolyte
Nature Protocols (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
