
Abstract
Crystal structures of G protein-coupled receptor (GPCR) ligand complexes allow a rational design of novel molecular probes and drugs. Here we report the structure-guided design, chemical synthesis and biological investigations of bivalent ligands for dopamine D2 receptor/neurotensin NTS1 receptor (D2R/NTS1R) heterodimers. The compounds of types 1–3 consist of three different D2R pharmacophores bound to an affinity-generating lipophilic appendage, a polyethylene glycol-based linker and the NTS1R agonist NT(8-13). The bivalent ligands show binding affinity in the picomolar range for cells coexpressing both GPCRs and unprecedented selectivity (up to three orders of magnitude), compared with cells that only express D2Rs. A functional switch is observed for the bivalent ligands 3b,c inhibiting cAMP formation in cells singly expressing D2Rs but stimulating cAMP accumulation in D2R/NTS1R-coexpressing cells. Moreover, the newly synthesized bivalent ligands show a strong, predominantly NTS1R-mediated β-arrestin-2 recruitment at the D2R/NTS1R-coexpressing cells.
Similar content being viewed by others

Introduction
Gprotein-coupled receptors (GPCRs) form the largest family of membrane proteins1. Because of their diversity and critical involvement in numerous cellular signalling processes in both central nervous system (CNS) and periphery, GPCRs represent today’s most popular drug targets attracting interdisciplinary scientific attention. Consequently, large progress has been made in understanding GPCR structures and modes of function. A growing number of studies showed that GPCRs not only exist as isolated entities but also interact within the plasma membrane by forming receptor dimers or higher-order oligomers2,3,4,5. Besides enabling cross-talk between individual signalling networks, receptor dimerization can induce activation of alternative signalling pathways6,7, influence ligand pharmacology and is critical for receptor trafficking and function3.
Dopamine D2 receptors (D2Rs), which belong to the family A of GPCRs, regulate a large number of physiological functions and are involved in a number of neuropsychiatric disorders including schizophrenia and Parkinson’s disease. Along with numerous other GPCRs, D2Rs have been proven to form homodimers8,9 and heterodimers10,11,12,13,14, and growing evidence indicates that altered D2R cooperativity may significantly contribute to CNS disorders15,16. Among receptors interacting with D2Rs in the CNS, the neurotensin receptor subtype 1 (NTS1R) together with its endogenous ligand, the tridecapeptide neurotensin, has gained substantial interest over the past decades. Both GPCRs are closely associated and highly co-localized in vivo17. For example, more than 80% of dopaminergic neurons in the mesolimbic system express NTS1R (ref. 18). Moreover, neurotensin was found to decrease the D2R-affinity for dopamine and other agonists in striatal19 and co-transfected HEK 293 T membrane preparations20. Evidence for the physical intramembrane interaction of both receptors was also conferred by means of bioluminescence resonance energy transfer, co-immunoprecipitation and attenuation of dopaminergic signalling in co-transfected human cells20,21. Since central administration of the neuropeptide in animals can mimic the effects of neuroleptic treatment, neurotensin has been hypothesized to act as endogenous antipsychotic22.
Powerful tools for studying GPCR dimerization are bivalent ligands consisting of two pharmacophores tethered by an appropriate linker23,24. Bivalent ligands bridging the proximate orthosteric-binding sites of a dimer provide valuable insights into the quaternary structure of receptor dimers and the functional relevance of GPCR dimerization. Because of their selective recognition properties, bivalent ligands can be used for a tissue-specific targeting of cells expressing an individual GPCR dimer. Pioneering work in this field was performed developing dimer-preferring ligands to investigate opioid receptor dimerization in vitro and in vivo25,26,27 and further compounds were synthesized to target GPCR homo- and heterodimers28,29,30,31,32,33. In theory, bivalent ligands successfully bridging two binding sites of adjacent protomers should confer extremely high affinity (resulting from the total binding energy of two recognition elements) and thus selectivity for the heterodimer. Most of the previous reports have shown compounds with only modest preference for heterodimers over monomers.
High-resolution crystal structures of GPCR-ligand complexes open new opportunities for the design of bivalent ligands. A carefully designed bivalent ligand bridging two neighboured receptor protomers should exhibit extremely high binding affinity. This approach should lead to high tissue selectivity between heterodimer-expressing cells and those that express only one individual receptor6. Our work presents heterobivalent D2R/NTS1R ligands of type 1-3 comprising NT(8-13), the active fragment of the neuropeptide neurotensin, covalently linked to three different D2R-specific pharmacophores. These newly synthesized bivalent compounds exhibit high selectivity up to three orders of magnitude and picomolar Ki values in D2R/NTS1R-coexpressing cells compared with cells expressing D2R only. Using bivalent ligands containing an agonist D2R pharmacophore substructure, we demonstrate that Gi/Go-promoted D2R signalling is attenuated in the D2R/NTS1R coexpressing cells, while the compounds behave as full dopamine receptor agonists in cells singly expressing D2R.
Results
Design
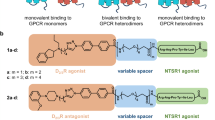
To design heterobivalent ligands, we intended to connect three different D2R pharmacophores to the NTS1R agonist NT(8-13) via an affinity-generating biphenyltriazole-moiety (lipophilic appendage)34,35 and ω-amino acid-functionalized polyethylene glycol (PEG) spacers (Fig. 1). As D2R pharmacophores, we used the D2R/D3R antagonist eticlopride, co-crystallized in complex with D3R (ref. 36), the privileged structure of a phenylpiperazine-based scaffold37 and an aminoindane-type agonist32. Suitable attachment points for the connection of the pharmacophores with the linker were identified using the crystal structures of NTS1R (refs 38, 39) and D3R (ref. 36). Inspection of the crystal structures revealed that the N-terminus of NT(8-13) and the 4’-position of eticlopride are accessible from the extracellular side. In an effort to determine a suitable linker length, we generated a D2R/NTS1R heterodimer model (Fig. 2) consisting of a D2R homology model40 (which was based on the D3R crystal structure) and the NTS1R crystal structure39 (Supplementary Note 1; Supplementary Tables 1,2; and Supplementary Fig. 1). As templates to build the dimer model, we considered 16 crystal structures of 12 different GPCRs displaying homodimers with 18 individual receptor orientations. We generated dimer models based on every template. Models were not considered further if they showed substantial clashes between the two receptors, as well as models revealing a high distance between the protomers or showing a low parallelism of the two protomers. Showing relatively high sequence similarity with D2R, the structure of a β1-adrenergic receptor (β1-AR) homodimer41 was selected as a template for the generation of the heterodimer model. The crystal structure revealed a dimer interface involving transmembrane helix 1 (TM1), TM2 and helix 8 (H8) that was previously reported to be important for D2R dimerization8 and validated by crosslinking studies at β1-AR (ref. 41). The model showed a minimum distance (beeline) of 42 Å between the attachment points of eticlopride and NT(8-13) (Fig. 2 and Supplementary Note 2). However, docking of eticlopride with the affinity-generating biphenyltriazole-moiety into the heterodimer model revealed two reasons why a longer spacer length should be required. First, the binding pocket of D2R restricts the D2R-attachment in a position not facing straight towards NTS1R (Supplementary Fig. 2a) and in addition, the way is partially blocked by the extracellular loop 1 of D2R and the N-terminus of NTS1R, resulting in a total distance of ∼55 Å. We concluded that at least two PEG-units, in addition to the biphenyltriazole-based attachment, should be necessary to enable a bivalent-binding mode (ligand 1b), while a ligand containing only one PEG-unit (ligand 1a, corresponding to a maximal linker length of ∼46 Å) should lack the ability to bridge the two binding sites and could thus serve as a control agent (Supplementary Fig. 2b). To determine an optimum linker length, we additionally designed compounds 1c and 1d bearing three and four units of the functionalized PEG-spacer, respectively. Using an identical approach, we designed the bivalent compounds of types 2 and 3 featuring phenylpiperazine- and aminoindane-based D2R pharmacophores. Here, the attachment points at the pharmacophores were identified based on docking studies (Supplementary Note 2 and Supplementary Fig. 3). We performed molecular dynamics (MD) on the bivalent ligands 1b, 2b and 3b (400 ns for each compound) in complex with the generated heterodimer model, because flaws in the process of ligand design would potentially appear as instabilities in the simulation systems. All three systems adopted stable receptor–ligand complexes during this time (Supplementary Note 3; Supplementary Fig. 4; and Supplementary Data 1, 2, 3).

Type 1-3 ligands contain NT(8-13) as NTS1R pharmacophore and differ in their D2R recognition element (type 1: eticlopride, antagonist; type 2: 2-methoxyphenylpiperazine, antagonist; and type 3: aminoindane, agonist). The spacer length connecting both pharmacophores ranges from 22 to 88 atoms (m=1–4). For bivalent control compounds 2e/f and 3e/f, tyrosine was replaced by N-homotyrosine (NhTyr).

On the basis of X-ray crystal structures, a D2R/NTS1R heterodimer model was generated and exploited for the design of bivalent ligands. A side view of dimer model is displayed at the bottom half and the ligand structures at the top of the figure. The middle part shows a magnified top view of the dimer model. Ribbons and surfaces of D2R and NTS1R are coloured in grey and red, respectively. Eticlopride (green) and NT(8-13) (blue) were positioned according to their coordinates in the crystal structures of D3R and NTS1R, respectively. Clearly, the N-terminus of NT(8-13) and the 4’-position of eticlopride are accessible from the extracellular side and were therefore selected as attachment points (highlighted as pink spots in all three representations). The beeline and hence the minimum distance connecting these two attachment points measures 42 Å.
Synthesis
Chemical synthesis was conducted on solid phase involving the generation of the peptidic sequence, followed by ligation of the individual linker and coupling with the carboxylate-functionalized dopaminergic pharmacophore. The affinity-generating biphenyltriazole-moieties were installed using click chemistry. To generate appropriate control agents, we linked the dopaminergic building blocks to a peptide–peptoid hybrid of NT(8-13) (ref. 42), which is highly similar to NT(8-13) but shows only poor NTS1R affinity (compounds 2e,f, 3e,f; Fig. 1 and Supplementary Table 3).
Radio-ligand binding
Binding profiles of the bivalent ligands of types 1–3 were determined by displacement of the radio-ligand [3H]spiperone from the human D2R in membranes from HEK 293 T cells singly expressing the D2R and in D2R/NTS1R-coexpressing cells (Table 1). Test compounds 1a, 2a and 3a containing biphenyltriazole-substituted eticlopride, phenylpiperazine and aminoindane moieties, respectively, linked to NT(8-13) by a short 22-atom spacer to NT(8-13) showed acceptable binding affinities to the D2R with Ki values ranging from 1.4 and 1.7 nM for 1a up to double-digit nanomolar values for 2a and 3a, in D2R and D2R/NTS1R expressing membranes (Table 1 and Fig. 3a–c). Extension of the linker to 44 atoms resulted in comparable affinities for 1b, 2b and 3b at the D2R monoexpressing cells, (Ki 9.9, 42 and 36 nM). However, binding characteristics at the D2R/NTS1R-coexpressing cells were changed dramatically by the elongation of the linker. Thus, we observed biphasic competition curves with two individual values for Ki high and Ki low (Fig. 3d–f). For all three compounds, high-affinity binding was observed at subnanomolar concentrations (Ki high 0.11–0.47 nM) with a high-affinity population of 31–55%, while the affinity for the low-affinity site ranged from 43 to 630 nM (Table 1). We suggest that the high-affinity Ki values represent a bivalent receptor-bridging binding mode of 1b, 2b and 3b to D2R/NTS1R heterodimers, whereas low-affinity Ki values reflect a monovalent-binding mode to D2R as a monomer or within a homo-/heterodimer. Thus, these newly designed ligands exhibit a 76–200-fold preference for the high-affinity bivalent interaction with the D2R/NTS1R heterodimer over monovalent-binding modes to D2R monoexpressing membranes.

Dopamine receptor binding of the bivalent ligands 1a–d, 2a–d and 3a–d was measured by displacement of the radio-ligand [3H]spiperone from membranes of HEK 293 T cells coexpressing D2R/NTS1R (blue open circles) or monoexpressing D2R only (grey filled circles). (a-c) Bivalent ligands with a spacer length of 22 atoms (m=1) result in monophasic competition-binding curves. (d–l) Biphasic-binding curves indicating bivalent ligand binding are observed when the linker length is increased to 44, 66 or 88 atoms (m=2–4) at membranes coexpressing both target receptors (D2R/NTS1R) but not at membranes with D2R only. Data points represent the mean±s.e.m. of 3–22 independent experiments (see Table 1 for details), each performed in triplicate.
In contrast, the respective analogues 2e/f and 3e/f (spacer length 22 and 44 atoms), containing a peptide–peptoid hybrid with almost no affinity for NTS1R instead of the highly similar peptide NT(8-13), displayed monophasic-binding curves at both, D2R and D2R/NTS1R-coexpressing membranes (Ki 15-40 nM for D2R and Ki 22–110 nM for D2R/NTS1R; Supplementary Fig. 5a–d). Typical monophasic-binding curves were also observed for the monovalent analogues of types 2 and 3 ligands 2g and 3g for both expression systems (Ki 20 and 21 nM for 2g and 3g at D2R and Ki 42 and 42 nM for 2g and 3g at D2R/NTS1R, respectively; Supplementary Fig. 5e,f).
In an attempt to find an optimum linker length, we also investigated the binding behaviour of compounds 1c–3c and 1d–3d with a spacer length of 66 and 88 atoms, respectively. Whereas ligands 1c and 1d maintained one-digit nanomolar affinity (Ki 2.5 and 9.0 nM), elongation of the spacer led to a loss of binding affinity for types 2 and 3 ligands (Ki 140–520 nM) at D2R monoexpressing membranes. Nevertheless, the biphasic-binding profiles with separated high- and low-affinity sites at D2R/NTS1R-coexpressing membranes were preserved or even enhanced for ligands 1c–3c and 1d–3d (Ki high 0.087–2.6 nM, Ki low 120–1,800 nM, Fig. 3g–l). Within the entire set of compounds, ligand 2d (spacer length 88 atoms) displayed the outstanding affinity of 87 pM for the high-affinity binding site. Interestingly, the preference for the high-affinity binding site versus the affinity for D2R monoexpressing membranes was more pronounced for types 2 and 3 ligands based on phenylpiperazines or aminoindane as D2R pharmacophores (76–4,700-fold), compared with the eticlopride-based derivatives (3.5–90-fold), with comparable fractions of high-affinity binding sites (50–66% high-affinity fraction).
Additional binding assays were performed in the presence of an excess of NT(8-13) (1 μM), which should prevent a bivalent-binding mode of the test compounds to D2R/NTS1R-coexpressing membranes by displacing the NT(8-13) pharmacophore of the bivalent ligands from NTS1R. In fact, co-incubation prevented high-affinity binding, resulting in typical sigmoidal monophasic curves (Fig. 4a and Supplementary Fig. 6a). Detailed analyses revealed a slightly reduced D2R-affinity in the presence of the monovalent NTS1R agonist, which is in agreement with the reduced Ki low observed for bivalent ligands at D2R/NTS1R-coexpressing membranes compared with D2R monoexpression (Table 1). Importantly, these findings are consistent with earlier studies demonstrating a negative effect of neurotensin especially on D2R agonist affinity19,20. The binding properties of the reference antagonist spiperone remained almost constant under these conditions (Ki 0.073 and 0.080 nM, n=3, in the absence and presence of 1 μM NT(8-13), respectively).

(a) Dopamine receptor binding of 3b (m=2, 44-atom spacer) at D2R/NTS1R in the absence (blue open circles) or presence (blue inverted triangles) of 1 μM NT(8-13). Incubation with the monovalent NTS1R agonist NT(8-13) prevents a bivalent binding mode and converts the biphasic-binding curve (Ki high 0.47±0.14 nM, Ki low 300±40 nM, n=22) into a monophasic sigmoid competition curve (Ki 63±8 nM, n=6). (b) Neurotensin receptor binding of 3b (m=2) at D2R/NTS1R in the absence (blue open circles) or presence (blue filled triangles) of 1 μM haloperidol. Incubation with the monovalent D2R antagonist prevents the bivalent binding mode, observed for the coexpression of D2R/NTS1R (Ki high 0.11±0.05 pM, Ki low 3.7±1.4 nM, n=9 versus Ki 0.79±0.21 nM, n=9). (c) When radio-ligand displacement studies were performed with striatal membranes and [3H]spiperone, biphasic-binding behaviour was observed for the bivalent ligand 3b (blue filled squares, Ki high 2.8±1.1 nM, Ki low 310±90 nM, fraction high-affinity sites 38±5%, n=5) alone, but not in the presence of 1 μM NT(8-13) (blue open sqares, Ki 28±3 nM, n=3). (d) For the bivalent control compound 3f comprising a peptoid-peptide hybrid instead of the NT(8-13) pharmacophore monophasic competition curves were observed in the absence (grey filled squares, Ki 28±16 nM, n=5) and presence (grey open squares, Ki 29±5 nM, n=4) of 1 μM NT(8-13). Data represent mean±s.e.m. of n independent experiments, each performed in triplicate.
However, even in the absence of NT(8-13), slight differences between the affinity for D2R monoexpressing membranes and the low-affinity binding site of D2R/NTS1R-coexpressing membranes were observed, suggesting that more complex ligand/receptor interactions might take place in the coexpressing membranes. Moreover, the simultaneous presence of at least three binding modes (bivalent and monovalent to D2R/NTS1R heterodimer, monovalent to D2R monomer) should putatively result in triphasic-binding curves, which we have not been able to resolve.
Above described experiments were performed with a twofold excess of NTS1R, concluding that most of [3H]spiperone-bound D2R were able to form D2R/NTS1R heterodimers. By changing the ratio towards a two- or three-fold excess of D2R, the high-affinity fraction, which corresponds to the bound receptor heterodimer, was rightward shifted and the biphasic character of the curve was gradually diminished (Supplementary Fig. 6b). Thus, a correlation between the ratio of protomers and the formation of molecular entities bound by bivalent ligands with particularly high affinity could be demonstrated.
Further competition experiments with the bivalent ligands 2b and 3b were conducted in presence of the non-hydrolysable GTP analogue GppNHp, thereby destabilizing receptor-G protein association. In fact, co-incubation with 100 μM GppNHp had no influence on the binding behaviour of the bivalent ligand 2b at membranes from D2R-expressing cells (Ki – GppNHp 42±5 nM versus Ki+GppNHp 45±6 nM). In contrast, a slight rightward shift of the Ki was observed for compound 3b, which is in good agreement with its D2R agonist pharmacophore (Ki – GppNHp 36±9 nM versus Ki+GppNHp 68±8 nM). However, at D2R/NTS1R-coexpressing membranes, a rightward shift of the high-affinity binding site occurred for both compounds (5.2- and 6.4-fold for 2b and 3b, respectively, Supplementary Fig. 7a,b). These changes are expected, since the agonist NT(8-13) as NTS1R-recognizing fragment is part of both bivalent ligands, and agonist affinity is strongly dependent on the presence of G proteins. Nonetheless, in the presence of GppNHp, the biphasic-binding behaviour is retained, leading to a 330- and 190-fold preference for the high- over the low-affinity binding site in D2R/NTS1R-coexpressing membranes.
To confirm the bivalent receptor-bridging binding mode, we performed reciprocal competition experiments by labelling the NTS1R with the radio-ligand [3H]neurotensin. Therefore, we used a homogenate with a 2.5-fold excess of D2R. Employing 3b, we observed a biphasic-binding curve in cells expressing the D2R/NTS1R heterodimer with a Ki high value of 0.11 pM and a Ki low at 1.7 nM, which was shifted to a monophasic sigmoidal binding curve in the presence of haloperidol (Ki 0.79 nM, Fig. 4b). Hence, incubation with the monovalent D2R antagonist efficiently prevented the bivalent-binding mode. Affinities for this competition-enforced monovalent-binding mode were found to be in good agreement with results obtained with membranes from CHO-cells stably expressing NTS1R only (Ki 0.86 nM; Supplementary Table 3 and Supplementary Methods).
To complement the results obtained with overexpressing heterologous cell lines with results from native brain tissue, competition-binding studies with [3H]spiperone and the bivalent ligand 3b in comparison with the control agent 3f (both with a spacer length of 44 atoms) were performed with membranes from porcine striatum. Convincingly, test compound 3b displayed a biphasic-binding behaviour with a 140-fold preference for the high-affinity binding site over the low-affinity receptor population (Ki high 2.8 nM, Ki low 310 nM, high-affinity fraction 38%). In good agreement with the results from heterologous cell lines, addition of 1 μM NT(8-13) reverted this biphasic-binding curve to a sigmoidal binding isotherm with a Ki value of 28 nM. In contrast, typical monophasic-binding curves with Ki values of 28 and 29 nM were observed in the absence and presence of 1 μM NT(8-13) for the highly similar peptide–peptoid hybrid ligand 3f, which proved to have almost no affinity for NTS1R (Fig. 4c,d). Thus, the connection of D2R- and NTS1R-addressing pharmacophores by an appropriate linker allows the superior recognition of heterodimers over monomers or homo(oligo-)mers not only in heterologous cell lines but also in native tissue. However, the observed preferences are less pronounced in striatal membranes, which might be, at least in part, explained by lower receptor expression levels leading to a lower propensity to form D2R/NTS1R heterodimers.
Functional evaluation
To measure activation profiles of the bivalent ligands 2b and 3b and their monovalent analogues 2g and 3g comprising the pharmacophore of a D2R antagonist and a D2R agonist, respectively, we performed a BRET-based cAMP accumulation assay43. Coupling to inhibitory Gαi/o proteins, the stimulation of the D2R leads to a decrease of cAMP, whereas activation of the Gαs-coupled NTS1R increases adenylyl cyclase activity. As expected, the reference agonist quinpirole potently inhibited forskolin-induced cAMP accumulation in cells expressing D2R only, while the phenylpiperazine-derived ligands 2b and 2g and the NTS1R-binding fragment NT(8-13) remained without significant effects (Fig. 5a).

Functional activity of the bivalent ligands 2b, 3b and their monovalent analogues 2g, 3g was determined in HEK 293 T cells coexpressing the cAMP-BRET biosensor CAMYEL and the D2R (a,d), the NTS1R (b,e) or both D2R and NTS1R (c,f). Cells were stimulated with increasing amounts of the ligands in the presence of 10 μM forskolin. cAMP production was normalized to the percentage of forskolin-induced cAMP concentration (100%). (a) While quinpirole potently inhibited cAMP formation, both D2R antagonists 2b, 2g and the NTS1R agonist NT(8-13) remained without significant effect on cells expressing D2R. (b) NTS1R could be stimulated by NT(8-13) and the bivalent ligand 2b, also bearing a NT(8-13) pharmacophore. (c) In the coexpressing cells, NT(8-13) induced an increase of cAMP, while quinpirole decreased the forskolin stimulated cAMP production. The bivalent ligand 2b also increased the cAMP production, similar to cells expressing NTS1R only. (d) Ligands 3b and 3g inhibited cAMP formation highly similar to quinpirole, revealing potent D2R agonism. (e) In cells monoexpressing NTS1R, only the bivalent ligand 3b stimulated receptor activation, while the monovalent analogue 3g caused no effect. (f) While the monovalent ligand 3g showed an effect similar to quinpirole, the bivalent ligand 3b induced further cAMP accumulation, indicating that its D2R-mediated effect was missing in the coexpressing cell line. Data represent mean±s.e.m. of 3–11 (for details see Supplementary Table 4) independent experiments each performed in triplicate; v=vehicle (PBS+10 μM forskolin).
Since we were unable to detect intrinsic activity for type 2 ligands in our cAMP accumulation assay, we tested the representative bivalent and monovalent ligands 2b and 2g for their capacity to prevent quinpirole-mediated inhibition of cAMP accumulation. As expected, both ligands were able to fully inhibit the effect of 10 nM quinpirole. In comparison, the type 1 ligand 1b and its pharmacologically active D2R fragment eticlopride were more potent and even showed an inverse agonist effect, leading to a 20–32% change in the basal cAMP level (Supplementary Fig. 8). The bivalent ligand 3b and the monovalent dopaminergic 3g bearing the aminoindane moiety displayed functional properties that were highly similar to quinpirole. Observed potencies (EC50) were in the low nanomolar range (2.3–5.0 nM), and maximum efficacies did not differ significantly among the three investigated D2R agonists (Fig. 5d and Supplementary Table 4). In cells expressing only NTS1R, neither the monovalent ligands 2g, 3g, nor quinpirole were able to exhibit receptor activation. However, the bivalent ligands 2b and 3b were as effective as NT(8-13), albeit at 10-fold higher concentrations (EC50 2.6 nM for NT(8-13) versus EC50 20.7 and 30.6 nM for 2b and 3b, Fig. 5b,e). In D2R/NTS1R-coexpressing cells, quinpirole and the monovalent D2R agonist 3g inhibited cAMP formation with similar potencies compared with cells monoexpressing D2R. The monovalent phenylpiperazine 2g had no effect on the intracellular cAMP concentration. Interestingly, all investigated bivalent ligands increased cytosolic cAMP in a similar manner as NT(8-13). Observed potencies were comparable to the monoexpressing NTS1R cells (EC50 2.0 nM for NT(8-13) versus 39.3 and 70.0 nM for 2b and 3b, respectively), although a slight loss in potency could be observed for the bivalent ligands (Fig. 5c,f). The extremely high affinity of the bivalent ligands could not be transduced into an increase in potency at D2R/NTS1R-coexpressing cells. When the spacer connecting the D2R and NTS1R pharmacophores was elongated to 66 atoms for the bivalent ligands 2c and 3c, similar observations concerning the activation of D2R, NTS1R and D2R/NTS1R heterodimers were made (Supplementary Fig. 9a–c and Supplementary Table 4).
The absence of D2R-mediated inhibition of cAMP accumulation in cells coexpressing D2R/NTS1R, is not specific for bivalent ligands, since a comparable attenuation of dopaminergic signalling is achieved when D2R/NTS1R-coexpressing cells are stimulated with quinpirole and NT(8-13) simultaneously (Supplementary Fig. 10a). In contrast, a reciprocal inhibition of NTS1R signalling by increasing concentrations of D2R agonist could not be observed (Supplementary Fig. 10b).
To exclude interference from forskolin stimulation or the relative receptor stoichiometry, experiments were also performed in the absence of forskolin and under conditions leading to enhanced NTS1R expression and therefore higher propensity to obtain D2R/NTS1R heterodimers. As illustrated in Supplementary Fig. 11a–e, these modifications did not result in significant changes of the receptor activation profiles of quinpirole, NT(8-13) or the bivalent ligands 2b and 3b. Coexpression of NTS1R and a signalling incompetent D2R-mutant (D80A)44,45 led to a loss of dopamine receptor signalling for the monovalent dopaminergic 3g and quinpirole while preserving the above described biphasic-binding behaviour and the activation profile of bivalent ligand 3b (Supplementary Fig. 12a,b) in D2R_D80A/NTS1R-coexpressing cells.
Besides G proteins, a class of adaptor proteins called β-arrestins are frequently found to interact with GPCRs. The recruitment of β-arrestin to a GPCR can lead to internalization but also initiate signalling events distinct from the G protein-mediated response46. To investigate the interaction of D2R/NTS1R heterodimers with β-arrestin-2, we made use of an assay system based on enzyme complementation (DiscoveRx PathHunter). Hence, HEK 293 cells stably expressing β-arrestin-2 fused to an enzyme acceptor (EA, galactosidase fragment) were transiently transfected with ProLink-tagged D2R together with or without cotransfection of wild-type NTS1R. Upon recruitment of β-arrestin-2 to D2R, the following enzyme complementation leads to conversion of a substrate and thereby chemiluminescence. The D2R agonist quinpirole induced β-arrestin-2 recruitment in D2R monoexpressing and D2R/NTS1R-coexpressing cells with similar potencies (EC50 55±3 versus 75±11 nM). Interestingly, stimulation with NT(8-13) induced β-arrestin-2 recruitment in the coexpressing cells, but not cells singly expressing D2R, indicating that β-arrestin-2 recruitment by NTS1R can be detected if it occurs in close proximity of D2Rs, as for example within a D2R/NTS1R heterodimer. Although the maximum effect remained below the response of quinpirole (77±3%), NT(8-13) elicited that response at 10-fold lower concentrations (EC50 7.5±2.1 nM; Fig. 6a,b). Application of an equimolar combination of both agonists led to an even enhanced efficiency of β-arrestin-2 recruitment (Emax 136±6%; Supplementary Fig. 13a).

β-Arrestin-2 recruitment was determined employing an assay based on enzyme complementation. HEK 293 cells stably expressing β-arrestin-2 tagged with the EA were transfected with ProLink-tagged D2R with (open blue circles) or without (filled grey circles) cotransfection of NTS1R (a) Quinpirole induces β-arrestin-2 recruitment in cells singly expressing D2R (n=9) and cells coexpressing D2R/NTS1R (n=11) with similar potencies. (b) NT(8-13) induces β-arrestin-2 recruitment with a maximum effect of 77±3% in cells coexpressing D2R/NTS1R (n=5), but not in D2R monoexpressing cells (n=4). (c) The phenylpiperazine-derived bivalent ligand 2b has no intrinsic activity in cells expressing D2R only (n=3), while it potently induces β-arrestin-2 recruitment in cells coexpressing D2R/NTS1R (n=7). Instead of a sigmoid curve, a bell-shaped dose–response profile is observed with a superior maximum effect as compared with both reference agonists. (d) The bivalent ligand 2a does not lead to β-arrestin-2 recruitment in D2R monoexpressing cells (n=3), but causes a typical sigmoid dose–response curve in the D2R/NTS1R-coexpressing cells (n=7). (e) The aminoindane-based bivalent ligand 3b induces β-arrestin-2 recruitment in cells expressing D2R (n=4) and cells coexpressing D2R/NTS1R (n=6). Coexpression of NTS1R leads to a significant increase in potency and efficacy and a bell-shaped dose–response curve as observed in c. (f) The aminoindane-type agonist with a 22-atom spacer (3a) leads to β-arrestin-2 recruitment in both types of transfected cells (n=3 for D2R and n=5 for D2R/NTS1R). Data represent mean±s.e.m. of n independent experiments, each performed in duplicate. Results were normalized to the maximum effect of quinpirole (100% for D2R and D2R/NTS1R).
For the bivalent ligands of the phenylpiperazine-type 2a and 2b with 22- and 44-atom spacers, no β-arrestin-2 recruitment was observed in cells expressing D2R only, which is in good agreement with the antagonist properties observed for 2a and 2b in the cAMP accumulation assay. Interestingly, a bell-shaped dose–response curve was observed for the bivalent ligand 2b in D2R/NTS1R-coexpressing cells. Maximum β-arrestin-2 recruitment was determined at a concentration of 300 nM (Emax 133%), while higher ligand concentrations led to an attenuated response (Fig. 6c). In contrast, a typical sigmoid dose–response curve was observed for the analogue 2a with the shorter 22-atom spacer (EC50 110±20 nM, Emax 88±4%, Fig. 6d). As expected, the bivalent ligands 3a and 3b bearing the aminoindane-type D2R agonist substructure elicited β-arrestin-2 recruitment in cells monoexpressing D2R (EC50 1,500±500 nM, Emax 87±5% and EC50 580±130 nM, Emax 82±1% for 3a and 3b respectively, Fig. 6e,f). Highly similar to the activation profiles of type 2 compounds, a bell-shaped dose–response curve with a maximum effect at a concentration of 300 nM to 1 μM (Emax 136%) was observed when NTS1R was coexpressed for 3b (44-atom spacer), but not 3a (22-atom spacer, EC50 37±8 nM, Emax 105±9%, Fig. 6e,f). Bell-shaped dose–response curves with enhanced efficacy were also observed for the bivalent ligands 2c and 3c (66-atom spacer; Supplementary Fig. 13b,c).
Additional experiments were performed employing a ProLink-tagged signalling incompetent D2R_D80A mutant coexpressed with NTS1R. Under these conditions, only NTS1R-mediated β-arrestin-2 recruitment can be detected. As expected, the NTS1R agonist NT(8-13), but not the D2R agonist quinpirole, was able to induce β-arrestin-2 recruitment in D2R_D80A/NTS1R-coexpressing cells. The potency of NT(8-13) was highly comparable to cells coexpressing wild-type D2R/NTS1R (EC50 5.9±1.2 nM for D2R_D80A/NTS1R and EC50 7.5±2.1 nM for wild-type D2R/NTS1R; Supplementary Fig. 14a,b). Highly similar to the results obtained with intact D2R, bivalent ligands with a short spacer (22-atoms, 2a, 3a) resulted in sigmoid dose–response curves (EC50 67±23 nM, Emax 98±4% and EC50 190±40 nM, Emax 113±7%, for 2a and 3a) in cells coexpressing NTS1R with the signalling incompetent D2R mutant, while bell-shaped curves with increased maximum efficacy were observed for the ligands with the longer spacer (2b,c, 3b,c, 44- and 66-atom spacer, Supplementary Fig. 14c–h). Ligands 2b,c and 3b,c reached maximum effects up to 210% relative to NT(8-13).
When the same experiments were performed in HEK 293 cells coexpressing NTS1R with ProLink-tagged protease-activated receptor subtype 2 (PAR2), only a very weak recruitment of β-arrestin-2 was observed for NT(8-13) and the representative bivalent ligands 2c and 3b (≤ 19%) compared with the PAR2 agonist f-LIGRLO-NH2 (ref. 47). The D2R agonist quinpirole was entirely inactive. Importantly, all dose–response curves showed a typical sigmoid profile (Supplementary Fig. 15a–d). These results indicate a specific effect of the bivalent ligands leading to bell-shaped dose–response curves in D2R/NTS1R-coexpressing cells.
Discussion
GPCR exist as monomers or cross-react forming dimers and higher-order oligomers. Because dimerization of GPCRs can result in modified ligand-binding and -signalling properties, a selective targeting of these entities is a powerful strategy in chemical biology and drug discovery. Irrespective of whether or not dimerization has physiological consequences per se, medicinal chemistry can take advantage of this phenomenon targeting drugs towards cells coexpressing an individual dimer-forming combination of GPCRs. In theory, bivalent ligands successfully bridging two binding sites of adjacent protomers should confer extremely high affinity (resulting from the total binding energy of two recognition elements) and thus selectivity for the receptor heterodimer. Most of the previous reports have shown compounds with only modest preference for heterodimers over monomers. In many cases, it has not been demonstrated that the two linked pharmacophores address two orthosteric-binding sites of two neighbouring protomers.
GPCR crystal structures may leverage an effective development of novel molecular probes and drug candidates48, because they can be used for structure-based in silico docking screens, giving access to new chemotypes and, as a consequence, to new biological profiles. Furthermore, they can guide the evolution of novel ligands by providing insights into attractive and repulsive ligand–receptor interactions and the relative topology of crucial moieties. Both strategies can be performed based on either the crystal structure of a given GPCR or starting from a homology model of a structurally highly similar congener. Using the co-crystallized ligands eticlopride and NT(8-13) as fragments for the design of bivalent ligands, the recently resolved X-ray crystal structures of NTS1R, D3R and a β1-AR dimer combined with homology modelling enabled us to determine the relative disposition of the pharmacophores to each other and to identify suitable attachment points for the spacer units. The strategy allowed a rational, structure-guided development of bivalent D2R/NTS1R ligands. The compounds 1b-d, 2b-d and 3b-d show unprecedented selectivity (up to three orders of magnitude) and binding affinity in the picomolar range for cells coexpressing both GPCRs, compared with cells that only express D2Rs. Preparations of porcine striatal membranes were used to investigate the biological relevance of our bivalent ligands. Although differences between high- and low-affinity binding sites were smaller, biphasic-binding curves confirmed a bivalent-binding behaviour.
A functional switch was observed for bivalent ligands containing a dopamine receptor agonist moiety. The compounds 3b and 3c behaved as agonists in cells singly expressing D2R inhibiting cAMP formation. However, no inhibitory effect on the NTS1R-promoted cAMP accumulation resulting from NTS1R activation by the NT(8-13) fragment was observed in D2R/NTS1R-coexpressing cells. Thus, the bivalent ligands 3b and 3c containing a D2R agonist pharmacophore behaved identical to bivalent ligands bearing a D2R antagonist moiety (2b and 2c), pointing towards a NTS1R-dominated signalling behaviour within D2R/NTS1R heterodimers. Yet, the exact molecular mechanism underlying this phenomenon is not fully understood. In particular, the extremely high binding affinity could not be translated into activation potency. The observed cAMP accumulation may be caused by monovalent binding to NTS1R, if the bivalently bound D2R/NTS1R heterodimer is unable to activate G proteins. However, interpretation of the functional experiments is far from trivial, as the overall response results from the activation of mixed populations of D2R and NTS1R monomers, homomers and heteromers. Moreover when dealing with bivalent ligands, at least three different (and probably even more) binding modes have to be considered: a monovalent-binding mode to each protomer as well as a bivalent, receptor-bridging binding mode.
As a second signalling pathway, we investigated the recruitment of β-arrestin-2. Employing an assay based on enzyme complementation, we could determine D2R-mediated β-arrestin-2 recruitment; while NTS1R-mediated engagement of β-arrestin-2 was only detectable in presence of the ProLink-tagged D2R. Compared with cells singly expressing D2Rs, coexpression and activation of NTS1R leads to a significant increase in potency. Thus, NTS1R protomer appears to dominate not only G protein coupling but also β-arrestin-2 recruitment in D2R/NTS1R-coexpressing cells. Importantly, bell-shaped dose–response curves were observed for the bivalent ligands 2b,c and 3b,c, whereas the structural analogues 2a and 3a with a shorter linker or a combination of two monovalent orthosteric ligands (quinpirole and NT(8-13)) showed regular sigmoid dose–response curves. In analogy to the binding behaviour of 2b,c and 3b,c (biphasic curves), the atypical dose–response relationship suggests a concentration-dependent contribution of different modes of receptor–ligand interactions. While it is not clear how exactly different binding modes influence the receptors’ capacity to recruit β-arrestin-2, the atypical dose–response curves obviously indicate that bivalent ligands with adequately designed spacer units display receptor activation characteristics distinct from monovalent ligands. Although the simultaneous presence of bivalent, receptor-bridging binding modes and monovalent-binding modes for bivalent ligands may represent a valid concept, we cannot exclude other, probably allosteric effects, leading to an altered binding and signalling behaviour.
Even though the exact molecular mechanism underlying the atypical functional behaviour remains to be elucidated, our study demonstrates the successful development of bi-orthosteric bivalent ligands targeting D2R/NTS1R heterodimers with unique properties. Because our target receptors are of major relevance for the pathophysiology of neurological and psychiatric disorders including Parkinson’s disease and schizophrenia, the D2R/NTS1R heterodimer may be a promising pharmacological target17. The tissue selectivity of bivalent D2R/NTS1R ligands may confer high potency and reduced side effects. Presumably, the in vivo bioavailability of our compounds of types 1–3 will not be suitable for their use as a drug. However, our newly developed bivalent ligands represent powerful pharmacological tools and may serve as a starting point for the development of innovative imaging agents and drugs addressing GPCR heterodimers, as sophisticated drug-delivery systems are currently developed.
Methods
Molecular modelling
D2R/NTS1R dimer models were generated by superimposing both our recently described homology model of the D2R (ref. 40) (which was based on the D3R crystal structure36) and the NTS1R crystal structure (PDB-ID 4BUO)39 with the so far resolved crystal structures of GPCR dimers. For details on the selection process see Supplementary Note 1 and Supplementary Tables 1 and 2. The final dimer model, created based on the crystal structure of the β1-AR dimer (PDB-ID 4GPO)41, was submitted to an energy minimization procedure as described previously40. Ligand positions were obtained in different ways. The coordinates of NT(8-13) in the crystal structure of NTS1R (ref. 39) were maintained for its position in the heterodimer model. The position of eticlopride was obtained by superimposing the crystal structure of D3R (ref. 36), including co-crystallized eticlopride, with our D2R homology model, followed by a transfer of the eticlopride coordinates to the D2R homology model. Coordinates of the remaining compounds were achieved by docking using AutoDock Vina49 as described previously40. Out of the 20 best-ranked conformations, one final conformation for each ligand was selected based on the scoring function of AutoDock Vina, experimental data and a manual inspection followed by an additional energy minimization. The all-atom force field ff99SB (ref. 50) was used for receptors and NT(8-13) and the general AMBER force field (GAFF)51 was used for the remaining ligands. A formal charge of +2 was assigned to NT(8-13), with the N-terminus and side chains of arginine protonated and the C-terminus deprotonated. A formal charge of +1 was assigned to the D2R ligands, here the basic nitrogen was protonated. Further details on heterodimer model generation, docking procedures and MD simulations are provided in Supplementary Notes 1–3; Supplementary Figs 1–4; and Supplementary Tables 1 and 2. Snapshots of MD simulations are provided as Supplementary Data 1, 2, 3. All figures were prepared using the UCSF Chimera package 1.10 (ref. 52).
Synthesis and characterization of bivalent ligands
Detailed schemes and conditions for the synthesis of the bivalent ligands 1a–d, 2a–f and 3a–f and the monovalent analogues 2g and 3g are provided in Supplementary Figs 16–18. Detailed methods and characterization for all compounds and precursors are provided as Supplementary Methods. For nuclear magnetic resonance analysis of the small molecules described in this article, see Supplementary Figs 19–34.
Cell culture
HEK 293 T cells (ATCC accession number CRL-11268) and HEK 293 cells stably expressing the EA-tagged β-arrestin-2 fusion protein (DiscoveRx) were maintained in DMEM/F-12 supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 μg ml−1 penicillin, 100 μg ml−1 streptomycin and 150 μg ml−1 hygromycin for EA-β-arrestin-2 cells at 37 °C, 5% CO2 (all cell culture reagents purchased from Invitrogen/Thermo Fisher Scientific). Cell lines were tested for mycoplasma contamination using the MycoAlert Plus detection kit (Lonza, Verviers, Belgium) on a regular basis.
Receptor-binding experiments
Receptor-binding studies were carried out in analogy to a previously described method53. Accordingly, competition-binding experiments with the human D2LR were perfomed using preparations of membranes from HEK 293 T cells, which were transiently transfected with the D2LR (from Missouri S&T cDNA Resource Center (UMR), Rolla, MO) using the Mirus TransIT-293 transfection reagent (purchased from MoBiTec, Goettingen, Germany). The assays were carried out in binding buffer (50 mM Tris pH 7.4, 5 mM MgCl2, 1 mM EDTA, 100 μg ml−1 bacitracin and 100 μg ml−1 soybean trypsin inhibitor) at a final volume of 200 μl with a protein concentration of 5–8 μg per assay tube, KD values of 0.048–0.060 nM and corresponding Bmax values of 960–970 fmol mg−1, together with [3H]spiperone (specific activity 81 Ci mmol−1, PerkinElmer, Rodgau, Germany) at a final concentration of 0.20–0.25 nM. Binding experiments with the co-transfected receptors were performed using membrane preparations from HEK 293 T cells, which were transiently transfected (Mirus TransIT-293) with the D2LR and NTS1R (from UMR) in the appropriate ratio of cDNA. Competition-binding experiments with the resulting homogenates of membranes coexpressing both receptors were carried out at a protein concentration of 1–8 μg per assay tube together with [3H]spiperone at a final concentration of 0.10–0.25 nM. For the detailed investigation of the heterobivalent ligands membranes with receptor densities of D2LR and NTS1R in the ratio of 1:2 (KD values of 0.053–0.080 nM, Bmax=800 fmol mg−1 for D2LR, 1,500–2,000 fmol mg−1 for NTS1R), 2:1 (KD 0.040 nM, Bmax=2,000 fmol mg−1 D2LR 1,000 fmol mg−1 NTS1R) and 3:1 (KD 0.050 nM, Bmax=8,000 fmol mg−1 D2LR, 2,500 fmol mg−1 NTS1R) were used. Competition-binding experiments with [3H]neurotensin (final concentratrion 0.50 nM, specific activity 101 Ci mmol−1; PerkinElmer, Rodgau, Germany) were performed at a protein concentration of 2 μg per assay tube and relative expression levels of 2.5:1 (Bmax=3,000 fmol mg−1 D2LR, 1,200 fmol mg−1 NTS1R, KD 0.50 nM). Unspecific binding was determined in the presence of haloperidol (10 μM for D2LR) or NT(8-13) (10 μM for NTS1R). Protein concentration was established by the method of Lowry using bovine serum albumin as standard54.
Binding experiments with porcine striatal membranes were performed as described above together with [3H]spiperone (final concentration 0.20–0.24 nM) at a protein concentration of 20 μg per tube (Bmax=220 fmol mg−1 D2LR, 140 fmol mg−1 NTS1R, KD=0.090 nM).
Data analysis
The resulting competition curves of the receptor-binding experiments were analysed by nonlinear regression using the algorithms in PRISM 6.0 (GraphPad Software, San Diego, CA). For each individual experiment, the data were fitted using a monophasic competition model to provide an IC50 value, which was then transformed into a Ki value according to the equation of Cheng and Prusoff55. The monophasic fit was accepted, unless a biphasic competition model providing two individual values for Ki high and Ki low resulted in a statistically significant better fitting of the data (extra sum-of-squares F-test, P<0.05).
cAMP BRET Assay
HEK 293 T cells were transiently transfected with pcDNA3L-His-CAMYEL (ref. 43) (ATCC) and D2SR and/or NTS1R at a cDNA ratio of 2:2 or 2:2:2 (unless indicated otherwise), respectively, using Mirus TransIT-293 transfection reagent. Resulting receptor expression levels were determined in saturation-binding experiments with membranes from transfected HEK 293 T cells and found to be 21±6 pmol mg−1 protein for D2SR monoexpression, 25±13 pmol mg−1 for NTS1R monoexpression and 15±4 and 7.2±1.7 pmol mg−1 for D2SR and NTS1R (1:1 transfection ratio) or 5.6±3.0 and 26±11 pmol mg−1 for D2SR and NTS1R (0.5:3.5 transfection ratio), respectively in the coexpressing cells. 24 h post-transfection cells were seeded into white half-area 96-well plates at 2.0 × 104 cells per well and grown overnight. On the following day phenol red free medium was removed and replaced by PBS and cells were serum starved for 1 h before treatment. The assay was started by adding 10 μl coelenterazine-h (Promega, Mannheim, Germany) to each well to yield a final concentration of 5 μM. After 5 min incubation, compounds were added in PBS containing 50 μM forskolin (final concentration 10 μM). Reads of the plates started 15 min after agonist addition. BRET readings were collected using a CLARIOstar plate reader (BMG LabTech, Ortenberg, Germany). Emission signals from Renilla Luciferase and YFP were measured simultaneously using a BRET1 filter set (475–30 nm/535–30 nm). BRET ratios (emission at 535–30 nm/emission at 475–30 nm) were calculated and dose–response curves were fitted by nonlinear regression using the algorithms of PRISM 6.0. Curves were normalized to basal BRET ratio obtained from dPBS (0%) and the effect of 10 μM forskolin (100%).
β-Arrestin-2 recruitment assay
The measurement of β-arrestin-2 recruitment stimulated by receptor activation was performed by utilizing the PathHunter assay purchased from DiscoveRx (Birmingham, UK) according to the manufacturer’s protocol. Accordingly, HEK 293 cells stably expressing the EA-tagged β-arrestin-2 fusion protein (provided by DiscoveRx) were transiently transfected with the ProLink(ARMS2-PK2)-tagged D2SR (or the respective ARMS2-PK2-tagged D2SR_D80A mutant) together with or without cotransfection of NTS1R at a cDNA ratio of 1:3 using Mirus TransIT-293 transfection reagent. Resulting receptor expression levels were determined in saturation-binding expriments with membranes from the same pool of cells and found to be 3.5±0.9 pmol mg−1 protein for D2SR and 4.2±0.3 pmol mg−1 for NTS1R in the D2R/NTS1R coexpression and 3.5±1.2 and 11.5±3.3 pmol mg−1 for D2SR_D80A and NTS1R, respectively for coexpression of the signalling incompetent D2R mutant together with NTS1R. 24 h after transfection, cells were detached using Versene (Invitrogen), 5,000 cells per well were seeded in 384-well plates and maintained at 37 °C, 5% CO2 for 24 h. After incubation with different concentrations of test compounds (from 10−15 to 10−4 M final concentration) in duplicates for 5 h, the detection mix was added and incubation was continued for further 60 min. Chemiluminescence was determined with a CLARIOstar reader for microplates (BMG LabTech). Resulting responses were normalized to the maximum effect obtained with quinpirole (100%) and the basal response (vehicle, 0%), or relative to the response of NT(8-13) (100%) when the signalling incompetent D2SR_D80A mutant was used. Dose–response curves were calculated by nonlinear regression using the algorithms of PRISM 6.0. Control experiments were performed using the same approach and transfection of ProLink(PK1)-tagged PAR2 together with wild-type NTS1R. Under these conditions, NTS1R expression levels were determined to be 8.7±1.2 pmol mg−1 protein. Resulting responses were then normalized to the effect of the PAR2 agonist f-LIGRLO-NH2.
Data availability
The data that support the findings of this study are available within the Supplementary Information files and/or from the corresponding authors upon request.
Additional information
How to cite this article: Hübner, H. et al. Structure-guided development of heterodimer-selective GPCR ligands. Nat. Commun. 7:12298 doi: 10.1038/ncomms12298 (2016).
References
Kobilka, B. K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta 1768, 794–807 (2007).
Bouvier, M. Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci. 2, 274–286 (2001).
Ferre, S., Ciruela, F., Woods, A. S., Lluis, C. & Franco, R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 30, 440–446 (2007).
George, S. R., O'Dowd, B. F. & Lee, S. P. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Discov. 1, 808–820 (2002).
Han, Y., Moreira, I. S., Urizar, E., Weinstein, H. & Javitch, J. A. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat. Chem. Biol. 5, 688–695 (2009).
Bellot, M. et al. Dual agonist occupancy of AT1-R-alpha2C-AR heterodimers results in atypical Gs-PKA signaling. Nat. Chem. Biol. 11, 271–279 (2015).
Milligan, G. G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br. J. Pharmacol. 158, 5–14 (2009).
Guo, W. et al. Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 27, 2293–2304 (2008).
Albizu, L. et al. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 6, 587–594 (2010).
Scarselli, M. et al. D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J. Biol. Chem. 276, 30308–30314 (2001).
Lee, S. Oligomerization of dopamine and serotonin receptors. Neuropsychopharmacology. 23, S32–S40 (2000).
Hillion, J. et al. Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J. Biol. Chem. 277, 18091–18097 (2002).
Urizar, E. et al. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat. Chem. Biol. 7, 624–630 (2011).
Hasbi, A. et al. Calcium signaling cascade links dopamine D1–D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl Acad. Sci. USA 106, 21377–21382 (2009).
Wang, M. et al. Schizophrenia, amphetamine-induced sensitized state and acute amphetamine exposure all show a common alteration: increased dopamine D2 receptor dimerization. Mol. Brain 3, 25 (2010).
Bagalkot, T. R. et al. Chronic social defeat stress increases dopamine D2 receptor dimerization in the prefrontal cortex of adult mice. Neuroscience 311, 444–452 (2015).
Binder, E. B., Kinkead, B., Owens, M. J. & Nemeroff, C. B. Neurotensin and dopamine interactions. Pharmacol. Rev. 53, 453–486 (2001).
Boules, M., Fredrickson, P., Muehlmann, A. & Richelson, E. Elucidating the role of neurotensin in the pathophysiology and management of major mental disorders. Behav. Sci. 4, 125–153 (2014).
Fuxe, K. et al. Intramembrane interactions between neurotensin receptors and dopamine D2 receptors as a major mechanism for the neuroleptic-like action of neurotensin. Ann. NY Acad. Sci. 668, 186–204 (1992).
Koschatzky, S., Tschammer, N. & Gmeiner, P. Cross-receptor interactions between dopamine D2L and neurotensin NTS1 receptors modulate binding affinities of dopaminergics. ACS Chem. Neurosci. 2, 308–316 (2011).
Borroto-Escuela, D. O. et al. Dopamine D2 receptor signaling dynamics of dopamine D2-neurotensin 1 receptor heteromers. Biochem. Biophys. Res. Commun. 435, 140–146 (2013).
Kinkead, B., Binder, E. B. & Nemeroff, C. B. Does neurotensin mediate the effects of antipsychotic drugs? Biol. Psychiatry 46, 340–351 (1999).
Hiller, C., Kühhorn, J., Gmeiner, P. & Class, A. G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J. Med. Chem. 56, 6542–6559 (2013).
Shonberg, J., Scammells, P. J. & Capuano, B. Design strategies for bivalent ligands targeting GPCRs. ChemMedChem. 6, 963–974 (2011).
Waldhoer, M. et al. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl Acad. Sci. USA 102, 9050–9055 (2005).
Daniels, D. J. et al. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl Acad. Sci. USA 102, 19208–19213 (2005).
Akgün, E. et al. Inhibition of inflammatory and neuropathic pain by targeting a Mu opioid receptor/chemokine Receptor5 heteromer (MOR-CCR5). J. Med. Chem. 58, 8647–8657 (2015).
McRobb, F. M., Crosby, I. T., Yuriev, E., Lane, J. R. & Capuano, B. Homobivalent ligands of the atypical antipsychotic clozapine: design, synthesis, and pharmacological evaluation. J. Med. Chem. 55, 1622–1634 (2012).
Butini, S. et al. Discovery of bishomo(hetero)arylpiperazines as novel multifunctional ligands targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors. J. Med. Chem. 53, 4803–4807 (2010).
Gogoi, S. et al. Novel bivalent ligands for D2/D3 dopamine receptors: significant cooperative gain in D2 affinity and potency. ACS Med. Chem. Lett. 3, 991–996 (2012).
Soriano, A. et al. Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. J. Med. Chem. 52, 5590–5602 (2009).
Kühhorn, J. et al. Development of a bivalent dopamine D(2) receptor agonist. J. Med. Chem. 54, 7911–7919 (2011).
Jacobson, K. A., Xie, R., Young, L., Chang, L. & Liang, B. T. A novel pharmacological approach to treating cardiac ischemia: binary conjugates of A1 and A3 adenosine receptor agonists. J. Biol. Chem. 275, 30272–30279 (2000).
Tschammer, N. et al. Highly potent 5-aminotetrahydropyrazolopyridines: enantioselective dopamine D-3 receptor binding, functional selectivity, and analysis of receptor-ligand interactions. J. Med. Chem. 54, 2477–2491 (2011).
Dörfler, M., Tschammer, N., Hamperl, K., Hübner, H. & Gmeiner, P. Novel D3 selective dopaminergics incorporating enyne units as nonaromatic catechol bioisosteres: synthesis, bioactivity, and mutagenesis studies. J. Med. Chem. 51, 6829–6838 (2008).
Chien, E. Y. et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095 (2010).
Löber, S., Hübner, H., Tschammer, N. & Gmeiner, P. Recent advances in the search for D3- and D4-selective drugs: probes, models and candidates. Trends Pharmacol. Sci. 32, 148–157 (2011).
White, J. F. et al. Structure of the agonist-bound neurotensin receptor. Nature 490, 508–513 (2012).
Egloff, P. et al. Structure of signaling-competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli. Proc. Natl Acad. Sci. USA 111, E655–E662 (2014).
Hiller, C. et al. Functionally selective dopamine D2/D3 receptor agonists comprising an enyne moiety. J. Med. Chem. 56, 5130–5141 (2013).
Huang, J., Chen, S., Zhang, J. J. & Huang, X.-Y. Crystal structure of oligomeric β1-adrenergic G protein–coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 20, 419–425 (2013).
Einsiedel, J. et al. Discovery of highly potent and neurotensin receptor 2 selective neurotensin mimetics. J. Med. Chem. 54, 2915–2923 (2011).
Jiang, L. I. et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 282, 10576–10584 (2007).
Peterson, S. M. et al. Elucidation of G-protein and β-arrestin functional selectivity at the dopamine D2 receptor. Proc. Natl Acad. Sci. USA 112, 7097–7102 (2015).
Neve, K. A., Cox, B. A., Henningsen, R. A., Spanoyannis, A. & Neve, R. L. Pivotal role for aspartate-80 in the regulation of dopamine D2 receptor affinity for drugs and inhibition of adenylyl cyclase. Mol. Pharmacol. 39, 733–739 (1991).
Lefkowitz, R. J. A brief history of G protein-coupled receptors (Nobel Lecture). Angew Chem. Int. Ed. Engl. 52, 6366–6378 (2013).
Hollenberg, M. D. et al. Derivatized 2-furoyl-LIGRLO-amide, a versatile and selective probe for proteinase-activated receptor 2: binding and visualization. J. Pharmacol. Exp. Ther. 326, 453–462 (2008).
Shoichet, B. K. & Kobilka, B. K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 33, 268–272 (2012).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Hornak, V. et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 (2006).
Jojart, B. & Martinek, T. A. Performance of the general amber force field in modeling aqueous POPC membrane bilayers. J. Comput. Chem. 28, 2051–2058 (2007).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Hübner, H., Haubmann, C., Utz, W. & Gmeiner, P. Conjugated enynes as nonaromatic catechol bioisosteres: synthesis, binding experiments and computational studies of novel dopamine receptor agonists recognizing preferentially the D3 subtype. J. Med. Chem. 43, 756–762 (2000).
Lowry, O. H., Rosebrough, N. J., Farr, A. L. & Randall, R. J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275 (1951).
Cheng, Y.-C. & Prusoff, W. H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 (1973).
Acknowledgements
This work was supported by the German Research Foundation (DFG Gm 13/8, GRK 1910). We thank A. Christopoulos and R. Lane (Monash University) for helpful discussions.
Author information
Authors and Affiliations
Contributions
M.G., C.S and L.L. synthesized all compounds. H.H. performed and analysed the radio-ligand-binding assays and β-arrestin-2 recruitment assays. T.S. performed cAMP accumulation assays. J.K. performed molecular modelling. T.C. supervised molecular modelling. D.M. conceived pharmacological experiments, analysed the biological data and wrote the manuscript. P.G. was responsible for the overall project strategy, provided project supervision and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-34, Supplementary Tables 1-4, Supplementary Notes 1-3, Supplementary Methods and Supplementary References (PDF 4103 kb)
Supplementary Data 1
MD ligand 1b (TXT 387 kb)
Supplementary Data 2
MD ligand 2b (TXT 386 kb)
Supplementary Data 3
MD ligand 3b (TXT 386 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hübner, H., Schellhorn, T., Gienger, M. et al. Structure-guided development of heterodimer-selective GPCR ligands. Nat Commun 7, 12298 (2016). https://doi.org/10.1038/ncomms12298
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms12298
This article is cited by
-
Constrained catecholamines gain β2AR selectivity through allosteric effects on pocket dynamics
Nature Communications (2023)
-
Update on GPCR-based targets for the development of novel antidepressants
Molecular Psychiatry (2022)
-
Prediction of molecular interactions and physicochemical properties relevant for vasopressin V2 receptor antagonism
Journal of Molecular Modeling (2022)
-
Bivalent ligands promote endosomal trafficking of the dopamine D3 receptor-neurotensin receptor 1 heterodimer
Communications Biology (2021)
-
An allosteric modulator binds to a conformational hub in the β2 adrenergic receptor
Nature Chemical Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
