
Abstract
Yak domestication represents an important episode in the early human occupation of the high-altitude Qinghai-Tibet Plateau (QTP). The precise timing of domestication is debated and little is known about the underlying genetic changes that occurred during the process. Here we investigate genome variation of wild and domestic yaks. We detect signals of selection in 209 genes of domestic yaks, several of which relate to behaviour and tameness. We date yak domestication to 7,300 years before present (yr BP), most likely by nomadic people, and an estimated sixfold increase in yak population size by 3,600 yr BP. These dates coincide with two early human population expansions on the QTP during the early-Neolithic age and the late-Holocene, respectively. Our findings add to an understanding of yak domestication and its importance in the early human occupation of the QTP.
Similar content being viewed by others

Introduction
Domestication of livestock species was a key factor in triggering the socioeconomic transition in humans from a hunter–gatherer lifestyle to one of nomadic pastoralism or agricultural settlement1,2. This process occurred first in the Middle East ∼11,000 years ago and later in other parts of the world. The Qinghai-Tibet Plateau (QTP) is the world’s largest and highest plateau with an area of ∼2.5 million square kilometres and an average elevation of 4,200 m. Humans are known to have colonized this vast area of Asia by at least 20,000 years ago and subsequent large-scale human population expansions occurred during the early Neolithic (10,000–7,000 years before present (yr BP)) and late Holocene (4,000–3,000 yr BP)3,4,5. The bovine yak species is endemic to this region. Historical records and archaeological evidence suggest that yak pastoralist societies were established in the QTP by ∼4,500 yr BP (refs 6, 7) and previous analyses of mitochondrial DNA variation indicate that yaks were domesticated during the early Neolithic period, some time between 6,000 and 12,000 yr BP (refs 8, 9). Since then, yak has become the mainstay of Tibetan pastoral society and >14 million domestic yaks are currently kept on the QTP, providing food, shelter, fuel and transport for the indigenous human population10,11. The ancestral wild yak population is threatened, but still exists with regular gene flow occurring between wild and domestic populations (Supplementary Note 1). Because yak domestication preceded the development of a human pastoral lifestyle in the QTP, a plausible hypothesis is that yak domestication was closely associated with early human population expansion in the region.
To examine the domestication of yak in more detail, we compare the genomes of wild and domestic yaks and investigate genetic changes underlying domestication. We use coalescent modelling to date yak domestication and population expansions more precisely than can be inferred from available archaeological and palaeontological evidence, and relate our findings to information on the prehistoric development of human society on the QTP.
Results
Genome resequencing and genetic variation
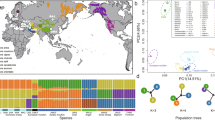
We analysed genome sequences from 13 wild yaks, representing three highly diverged mitochondrial lineages8,9, and 59 domestic yaks from different locations on the QTP (Fig. 1a) representing 48 animals from unselected landraces (D2 population) as well as 11 Tianzhu white yaks (D1 population), which since 130 years ago11,12 have been bred by strict selection of coat colour (Fig. 1b). Genome resequencing accomplished an average depth of 6.7 × and average genome coverage of 98% (Supplementary Table 1). We detected a total of 14.56 million high-quality single nucleotide polymorphisms (SNPs), most of which (76.4%) are located in intergenic regions (Supplementary Table 2).

(a) The geographic distribution of the sampling locations for wild (dark red) and domestic (dark blue) yaks. The coloured areas indicate geographic distribution of wild yaks (light red), Tianzhu county (green) and the QTP (grey). (b) A neighbour-joining phylogenetic tree constructed using whole-genome SNPs data. The scale bar represents level of similarity. W: wild yaks; D1, the Tianzhu white breed; and D2: all of the remaining domestic yaks. (c) Principal component (PC) analysis plots of the first two components. The fraction of the variance explained is 3.24% for PC1 and 1.36% for PC2, with Tracy–Widom P<10−44 (Supplementary Table 3). (d) Population structure plots with K=2–5. The y axis quantifies the proportion of the individual’s genome from inferred ancestral populations, and x axis shows the different populations. Geographic information is provided in Supplementary Table 1. (e) Decay of linkage disequilibrium of D1, D2 and W populations measured by r2.
Genetic changes underlying domestication
To examine the genome-wide relationships and divergence between wild and domestic yak populations, we visualized pairwise genetic distances in a neighbour-joining tree (Fig. 1b, consensus tree based on 1,000 bootstrap replicates shown in Supplementary Fig. 1). This revealed a clear split between wild and domestic yaks despite continuing gene flow between them, and also separation of Tianzhu white yaks within the domestic population (Supplementary Note 1). Principal component analysis as well as model-based clustering yielded similar results (Fig. 1c,d; Supplementary Note 1; and Supplementary Table 3). Domestication often reduces effective population size (Ne) and genetic diversity13,14; but we obtained similar sequence diversity (π) values of 0.0013 and 0.0014 for wild and domestic yaks, respectively (Supplementary Fig. 2 and Supplementary Table 4). We further found that the estimated population-differentiation statistic (FST) between wild and domestic yaks is only 0.058 (Supplementary Table 4), which is smaller than between taurine and zebu cattle or between diverged taurine cattle breeds15. FST estimates supported the gene flow occurring between wild and domestic yaks (Supplementary Note 1).
We detected genomic regions that have been subject to selection as inferred from high wild/domestic π log-ratios and an extreme divergence of allele frequencies of wild and domestic yaks16,17 (Fig. 2a and Supplementary Fig. 3). We identified 182 potential selective-sweep regions with an average size of 79.5 kb, together comprising around 14.5 Mb or 0.54% of the assembled genome. The role of these regions is confirmed by significantly lower values of Tajima’s D and higher linkage disequilibrium patterns (P values 2.7 × 10−12 and 1.5 × 10−4, respectively, Wilcoxon rank-sum test, Supplementary Note 2) in domestic populations. These regions harbour 209 annotated protein-coding genes (Supplementary Data 1), which are expected to represent targets of selection. Among these, GO group GO:0051969 (regulation of transmission of nerve impulse) was overrepresented (P<0.05, Supplementary Table 5 and Supplementary Data 1) with eight genes affecting synaptic circuitry and neurological processes (Arc, ASPA, ATP2B2, MYO6, NTRK2, Rab40c, SNCA and TG). From these genes and 30 other genes (Supplementary Data 1) involved in brain and neuronal development, 19 are considered to be associated with behaviour. ADCYAP1R1 (Fig. 2b) encodes a pituitary adenylate cyclase-activating polypeptide receptor that in humans is strongly expressed in the amygdala and hippocampus, and is associated with fear response, threat stimuli, post-traumatic stress disorder and other anxiety disorders. Adcyap1r1-deficient mice exhibit strongly reduced anxiety-like behaviour18. SCRIB (Fig. 2c) encodes the scribbled planar cell polarity protein, which is a key regulator of brain development and spine morphology. Scrib1 knockout mice exhibit enhanced learning and memory abilities and impaired social behaviour correlated with altered neuronal morphology19. PLXNB1 encodes a neuronal receptor for semaphorins and has an important role in developing nervous systems and controlling axon guidance20. A recent quantitative trait loci study in rat identified PLXNB1 as a candidate gene contributing to differences in tameness and aggression21, which are expected to be important during the early phase of animal domestication2. The pathways of brain and neuronal development identified here to be under selection during yak domestication are similar to those reported previously for rabbit22 and cat23, suggesting common features of domestication in these unrelated species.

(a) Distribution of ln ratio (θπ,wild/θπ,domestic) and FST of 50 kb windows with 10 kb steps. Red dots represent windows fulfilling the selected regions requirement (corresponding to Z test P<0.005, where FST≥0.17 and ln ratio≥0.65). Example of genes (b,c) with selection sweep signals in domestic yaks. FST, θπ and Tajima’s D values are plotted using a 5-kb sliding window. Wild (green) and domestic (blue) yaks are represented by different colours. Horizontal dashed lines represent mean whole-genome of corresponding values. Genes are shown at the bottom (black rectangle, coding sequences; red line, introns).
Only a few genes subject to selection were associated with specific physical characteristics or economically significant traits, such as TTLL1 and RHPN1 associated with sperm development and RHOD with early pregnancy. Also, a limited number of sweeps associated with coat colour were detected from an examination of genetic divergence between Tianzhu white yaks and other domestic yaks (Supplementary Note 3). In line with the low level of genetic and morphological differentiation recorded between wild and domestic yaks (Supplementary Table 4 and Supplementary Fig. 4), our analyses confirm that the effects of domestication in yaks are not as marked as for most other domestic species1,22. This may reflect a trade-off between survival of yaks in a harsh high-altitude environment and performance under pastoral conditions.
Demographic history
We employed the pairwise sequentially Markovian coalescent (PSMC) method24 to examine changes in effective population size (Ne) of the ancestral population of both wild and domestic yaks in response to Quaternary climatic change. We applied this method to our deep-coverage (>20 ×) yak genomes from three wild and four domestic yaks, including the reference genome. Both wild and domestic yaks exhibited similar demographic trajectories until about 20,000 years ago (Fig. 3a and Supplementary Fig. 5). The ancestral Ne of yaks shows a peak at ∼1 Myr ago followed by two distinct declines. The first decline occurred ∼0.9 Myr ago, coinciding with extensive glaciation during the mid-Pleistocene25, with three highly divergent mitochondrial lineages known to have survived this decline9. Other animal species such as giant panda and golden snub-nosed monkeys living in the southern and southeastern QTP also suffered during the same period decreases in effective population size26,27. The second decline involved at least a threefold decrease in Ne, and occurred ∼40,000 years ago coinciding with the last glaciation25.

(a) Demographic history inferred by PSMC. The period of the Xixiabangma Glaciation (XG, 1,170–800 thousand years ago, kya), Naynayxungla Glaciation (NG, 780–500 kya) and the last glacial maximum (LGM, ∼20 kya) are shaded in grey. (b) Schematic of demographic scenario modelled in Fastsimcoal2. The ancestral population is in grey, wild yak in brown and domestic yak in blue. The width shows the relative effective population size. The figures at the arrows indicate the average number of migrants per generation between wild and domestic yaks. The folded genome-wide SFS from 13 wild yaks (c) and 59 domestic yaks (d). Different colours represent data before (blue) and after (orange) impute filtering of sites for which the correlation of observed and imputed date was <0.9.
We used the joint site frequency spectrum (SFS) approach implemented in fastsimcoal2 (ref. 28) to simulate more recent demographic fluctuations. Thirty alternative models of historical divergence were fitted to the allele-frequency spectrum of domestic and wild yak populations, incorporating strict isolation, isolation-with-migration, bottlenecks and/or growth (Supplementary Fig. 6). A demographic model in which domestic and wild yaks diverged through a dynamic process involving population bottlenecks in both wild and domestic yaks and extensive post-domestication gene flow produced a significantly better fit than alternative models (Fig. 3b). The allele-frequency spectrum simulated with the best model was very close to the spectrum generated from real data (Supplementary Fig. 7), demonstrating the accuracy of the calculations. Thus in the best fitting model domestication of yaks occurred ∼7,300 yr BP, with a 95% confidence interval of 7,227–7,914 yr BP, slightly later than the domestication of many other livestock species (10,000–8,000 yr BP), but preceding the introduction of taurine cattle to China 5,400–4,700 yr BP (ref. 29).
Analyses of mitochondrial, Y-chromosomal and autosomal DNA data suggest that modern humans began colonizing the QTP ∼30,000 yr BP and that their population size expanded rapidly first between 10,000 and 7,000 yr BP and later between 4,000 and 3,000 yr BP (refs 5, 30, 31). However, archaeological and anthropological evidence indicates that the earliest agricultural settlements in the northeastern QTP were established 5,200 yr BP or later3,32. During the early-Neolithic age, the climate on the QTP was warmer than today25, which may have favored persistence of a hunter–gatherer population in the region. Our results suggest that the yak was domesticated by 7,300 yr BP and may have been triggered by and facilitated the first expansion of human population size on the QTP at this stage. Given the absence of agricultural settlements at this time, the first pastoralists were probably nomadic herders. A similar domestication by nomadic people in another extreme environment has been described for reindeer33.
Later in the Holocene agriculture was established on the QTP, for example, the introduction of barley cultivation 4,000–3,600 yr BP (ref. 30). This coincided with a second human population expansion3,4,5,30,32 despite the colder climate of the late Holocene25. Interestingly, our coalescence analyses revealed a sixfold increase in population size of the domestic yak (Ne, from 1,100 to 6,500) during the same period (3,600 yr BP, Fig. 3b), which might have resulted from the second human population expansion on the QTP following the introduction of agriculture or contributed to this second expansion by providing a reliable resource of food, hides and transportation. According to our coalescent analysis, ∼500 years ago the Ne of the wild yak population seriously declined from 21,200 to 1,700, which is consistent with a loss of low-frequency variants (Fig. 3c,d) and a lower genetic diversity in current wild yaks (Supplementary Fig. 2 and Supplementary Table 4). This possibly resulted from habitat loss due to increasing human activities.
Discussion
Despite low morphological divergence and continuing gene flow, we detected a clear genetic split between wild and domestic yaks. We found that the genomes of domestic yaks exhibit clear signatures of selection at genes that probably affect animal behaviour and tameness according to previous reports on other animals22,23. These findings suggest that parallel processes of evolution have occurred during the domestication of unrelated animals across different localities of the world. Our study further indicates that the yak is likely to have been domesticated before 7,000 years ago and that domestication was closely associated with the expansion of the human population on the QTP during the early Neolithic period31. Moreover, following the introduction of agriculture30, a further increase in the effective population size of domestic yaks later in the Holocene may have resulted from or contributed to causing a second human population expansion and the subsequent development of human society on the QTP during this period3,4,5,30,32
Methods
Sample collection and sequencing
A total of 84 individuals (15 wild yaks and 69 domestic yaks, Supplementary Table 1) were collected and sequenced, yielding a data set of genomes from 13 wild and 59 domestic yaks without close relatives and with <50% missing data. The wild samples were collected from corpses of wild yaks in the central Kokohili region, which were identified as wild yaks because of their long hair and large skeletons. Domestic yaks were sampled across the species main geographic distribution. Samples were collected under the supervision of ethical committees and permission was obtained when necessary. For each yak, genomic DNA was extracted from muscle samples using a standard phenol/chloroform extraction34. The quality and integrity of the extracted DNA was checked by measuring the A260/A280 ratio and by agarose gel electrophoresis. Paired-end sequencing libraries with an insert size of 500 bp were constructed according to the Illumina manufacturer’s instructions for sequencing on the Hiseq 2,000 platform. Sequencing and base calling were performed according to the standard Illumina protocols.
Sequence quality checking
Duplicate reads caused by base-calling and adaptor contamination were removed. Reads with (i) ≥10% unidentified nucleotides (N), (ii) with a phred quality ≤7 for >65% of the read length or with (iii) a stretch of >10 bp identical to the adaptor sequence with up to two mismatches were removed or corrected using a k-mer frequency-based methodology35. Reads were also trimmed if they had three consecutive bp with a phred quality of ≤13, and discarded if they were shorter than 45 bp.
Sex-linked scaffolds
We used Blastz (ref. 36) to perform whole-genome alignment of the yak and taurine cattle genomes and to identify yak sex chromosomes (downloaded from National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov, UMD3.1.1, GCA_000003055.4). All hits against the cattle sex chromosome were treated as sex-linked scaffolds. A total of 186 scaffolds with a combined size of ∼134 Mb were aligned to the cattle sex chromosome and omitted from subsequent analyses.
Read mapping
High-quality reads were aligned to the Bos grunniens reference genome37 and mitochondrial reference genome (accession number: JQ692071.1) using BWA-MEM (0.7.10-r789) with default parameters38. Sequence Alignment/Map (SAM) format files were imported to SAMtools (v0.1.19)39 for sorting and merging and Picard (http://broadinstitute.github.io/picard/, version 1.92) to assign read group information containing library, lane and sample identity. The Genome Analysis Toolkit (GATK, version 2.6–4-g3e5ff60)40 was used to perform local realignment of reads to enhance the alignments in the vicinity of indel polymorphisms. Realignment was performed with GATK in two steps. The first step used the RealignerTargetCreator to identify regions where realignment was needed, and the second step used IndelRealigner to realign the regions found in the first step, generating for each individual a realigned Binary sequence Alignment/Map (BAM) file.
Filtering alignments
We removed all alignments that were not of sufficiently high quality for SNP detection and subsequent analyses. Alignments to be removed were identified using the following stepwise protocol: (i) discard reads that do not map uniquely; (ii) only use reads for which a mate can be mapped; (iii) discard ‘bad’ reads with flag ≥255; (iv) discard bases with a quality <20; and (v) discard reads with a mapping quality <30. We also adjusted the quality scores around indels using SAMtools and removed the alignments that anchored short scaffolds of <2 kb.
Filtering sites
To minimize the influence of sequencing and mapping bias, the following site types were discarded: (i) sites with unbalanced quality scores as determined using Wilcoxon rank-sum test with threshold of P<10−5; (ii) sites with strand bias (P<10−5); (iii) sites with extremely low (<2 ×) or extremely high (>18 ×) coverage, both thresholds being defined after investigating the coverage distribution empirically; (iv) sites that failed the Hardy–Weinberg Equilibrium test and P<10−3, using SAMtools and BCFtools39; and (v) sites for which the available information derived from <90% of the sampled domestic and/or wild populations. The combined application of these filters left us with a data set comprising ∼2.2 Gb, representing 81.7% of the genome.
SNP and genotype calling
Variant discovery analysis was conducted at the population level for wild and domestic yak samples separately. We used the SAMtools model41 implemented in analysis of next generation sequencing data (ANGSD)42 to estimate genotype likelihoods and generated Beagle files. A maximum likelihood approach43 was then used to infer major/minor states based on the genotype likelihoods. Minor allele-frequency polarized by major/minor state was also estimated from the genotype likelihoods based on Kim’s method44. A likelihood ratio test statistic for the allele-frequency based on a χ2 distribution with one degree of freedom and a P-value threshold of 1 × 10-6 was used as an SNP discovery criterion. SNPs were retained only if they could be genotyped in at least 90% of the sampled individuals from both domestic and wild populations. This yielded a total of 14.6 million SNPs.
A two-step procedure implemented in ANGSD was used to estimate the SFS: (i) sample allele-frequency likelihood files (.saf) were generated using the option ‘–doSaf 1’, with ancestral state being assigned by a cattle genome45 (17.4 ×); (ii) the allele-frequency likelihood files were optimized using the realSFS (ref. 46) programme in order the estimate the SFS. Genotypes were called using the full set of genotype likelihoods data. Using the sample allele frequency as a prior for genotype frequencies under the assumption of Hardy–Weinberg equilibrium, we then computed the posterior probabilities of the genotypes at each site for each individual.
Relationships
To identify closely related individuals, the programme PLINK v1.07 (ref. 47) was used to obtain pairwise estimates of Identity-By-State (IBS) scores between all samples. One wild and seven domestic individuals were excluded due to their high pairwise genetic similarity with another sampled individual (IBS>0.9), leaving only unrelated samples for use in the downstream analyses. We also discarded one wild and three domestic individuals with >50% missing data (Supplementary Table 1).
Genome-wide identity scores
To visualize genetic relatedness between domestic and wild populations, we calculated for individual SNPs identity scores as the sum of the products of the frequencies of both alleles with the frequencies of the same allele in the reference genome. Identity scores for 50 kb windows along the genome were averaged over the SNPs within the window (Supplementary Fig. 4).
Population genetics analysis
After mapping sequencing data against the reference yak mitochondrial genome (accession number: JQ692071.1), only positions covered by a minimum number of three independent unique reads with base qualities of ≥30 were used to call the consensus sequences. Eighty yak mitochondrial sequences were generated and aligned together with 81 sequences from B. grunniens, one from Bos taurus, one from Bos indicus, one from Bison bison and one from Bison bonasus (see the labels of external branches on Supplementary Fig. 8 for accession numbers). We partitioned the alignment into six main regions: the D-loop, ribosomal RNA, tRNA and the first, second and third codon positions for Coding DNA Sequence (CDS). The initiation and termination codons and overlapping regions between CDSs were excluded. We also removed sites with missing genotypes in >10% of the sampled individuals. The best mutational model for each of the partitions was then selected using ModelGenerator v851 (ref. 48) with eight rate categories. The partitions and their corresponding mutational models were used for Bayesian phylogenetic inference with MrBayes v3.22 (ref. 49), running two analyses in parallel, each with four Markov Chain Monte Carlo (MCMC) chains. The final tree topology was recovered after a total of 100,000,000 generations, sampling 1 in every 1,000 generations after discarding the first 25% as burn-in. The s.d. of split frequencies was below 0.01 after 100,000,000 generations, indicating the convergence of the four chains to the stationary distribution. The resulting tree, as drawn with MEGA v5.0 (ref. 50), is shown in Supplementary Fig. 1. This tree topology is consistent with the presence of distinct phylogeographical patterns and multiple divergent lineages in yaks as determined in previous studies8,9 based on D-loop and mitochondrial genomic sequences from a wide range of samples.
For autosomal genome data, a neighbour-joining tree was constructed with PHYLIP v3.695 (http://evolution.genetics.washington.edu/phylip.html) using the matrix of pairwise genetic distances. The ancestral states of the SNPs were determined using the close relative of the yak, B. taurus45, as the outgroup. A second frequency tree (Supplementary Fig. 1) was generated based on 1,000 bootstrap replicates using the consensus module of PHYLIP. FigTree (http://tree.bio.ed.ac.uk/software/figtree/) and MEGA v5.0 were used to visualize the phylogenetic trees. Principal component analysis of the SNPs was performed using the smartpca programme in EIGENSOFT v5.0 (ref. 51). A Tracy–Widom test was used to determine the significance level of the eigenvectors.
Geographic distances in km between individuals were calculated via the formula: Distance=acos(sin(lat1 × π/180)sin(lat2 × π/180)+cos(lat1 × π/180)cos(lat2 × π/180)cos(lon2 × π/180−lon1 × π/180)) × 6378.135, in which lat1 and lat2 are latitudes in degrees of the two individuals and lon1 and lon2 their longitudes (Supplementary Fig. 9).
Genome-wide patterns of heterozygosity and neutrality tests
The nucleotide diversity (π), population-differentiation statistic (FST), Tajima’s D statistic and Watterson estimator (θw) were calculated using a sliding window approach (50 kb window sliding in 10 kb steps)52,53,54. To compensate for missing data and variations in the depth-of-coverage across the different genomes (4.5–8.8 × , average coverage 6.7 ×), an empirical Bayesian method was used to calculate the posterior probabilities for the sample frequency spectrum using a maximum likelihood estimate of the SFS as the prior. The method takes genotype uncertainty into account and is based directly on genotype likelihoods rather than called genotypes. Only genomic windows in which at least 80% of bases were covered were considered to avoid coverage-related bias, leaving 207,111 windows with an average SNPs number of 346 per-window (min 34, max 3,298).
Screening for selective sweeps
To identify genomic regions that may have been subject to selection during domestication, we combined the two domestic yak populations (D1 and D2) as a single domestic gene pool. We scanned the genome for regions with the highest differences in genetic diversity (π log-ratio wild/domestic) and extreme divergence in allele frequency between wild and domestic populations using a genome-wide sliding window strategy. More specifically, we calculated the sequence diversity statistics (π), and the population-differentiation statistic (FST) using a 50 kb window with a 10-kb step. The π log-ratio was calculated as ln(πW)−ln(πD), where πW and πD are the nucleotide diversity values for the wild and domestic yaks, respectively. At a significance level of P<0.005 (Z test, with π log-ratio >0.65 and FST>0.17, Fig. 2a and Supplementary Fig. 3), we identified a total of 182 potential selective-sweep regions (with an average size of 79.5 kb, range from 10 to 450 kb) overlapping with 209 candidate genes, used for subsequent analysis and discussion.
To test whether the candidate selective-sweep regions had an excess of singleton polymorphisms, we computed the Tajima’s D value for domestic yaks using the same sliding window approach. Regions under selective sweeps had very significantly lower values of Tajima’s D (P=2.7 × 10−12, Wilcoxon rank-sum test). In addition, pairwise r2 values showed that the candidate regions exhibited significantly extended linkage disequilibrium (P=1.5 × 10−4, Wilcoxon rank-sum test). These results confirm the occurrence of selective sweeps in the identified regions.
The impact of population structure to selection signal was tested by repeating the sweep detection by comparing the two domestic populations D1 and D2 separately to the wild population (W). For 196 (93.8%) of 209 genes selection signals were statistically significant in both domestic populations; relatively strong selection signals were evident for the other 13 candidate genes but did not reach the significance threshold (Supplementary Data 1).
Functional classification of GO categories was performed using the Blast2GO programme55. Enrichment analysis was performed and the χ2 test was used to calculate the statistical significance of enrichment. The P values were adjusted by FDR and the adjusted P value cut-off was 0.05.
Demographic history
We inferred a demographic history for B. grunniens by applying the Pairwise Sequentially Markovian Coalescence model24 to the complete diploid genome sequences, excluding sexual chromosomes/scaffolds. This method reconstructs the history of changes in population size over time using the distribution of the most recent common ancestor (tMRCA) between two alleles in an individual. PSMC has high false-negative rates at low depth, which leads to a systematic underestimation of true event times. To ensure the quality of consensus sequences, we sequenced three wild and three domestic yaks to a high coverage of 20 × . DNA was prepared and libraries were built using the protocols described above. Consensus sequences were obtained using SAMtools and divided into non-overlapping 100 bp bins. Bases of low sequencing depth (less than a third of the average depth) or high depth (twice the average depth) were masked. The analysis was performed using the following parameters: −N25 −t15 −r5 −p ‘4+25 × 2+4+6’. The mutation rate per generation per site was estimated as: μ=D × g/2 T where D is the observed frequency of pairwise differences between two species, T is the estimated divergence time and g is the estimated generation time for the two species. The estimated generation time (g) was set to 3 years and the estimated divergence time was set to 4.7 Myr based on a previous study on cattle and yak56. These values yielded an estimated mutation rate of 5.84 × 10−9 mutations per generation per site for the yak. PSMC modelling was done using a bootstrapping approach, with sampling performed 100 times to estimate the variance of the simulated results.
As PSMC inference is known to be inaccurate for recent datings, we also inferred the joint demographic histories of the wild and domestic yak using the flexible and robust simulation-based composite-likelihood approach implemented in the fastsimcoal2 programme28, which infers demographic parameters from the SFS. The analysis was performed for 13 wild samples and 59 domestic samples. To improve the genotype accuracy and infer missing genotypes, we used BEAGLE (ref. 57) to infer the haplotypes of wild and domestic individuals from previously estimated genotypes. After investigating the empirical minor allele frequency distributions, we inferred haplotypes for non-coding sites alone with estimated minor allele frequency values of >0.038 for wild yak and >0.008 for domestic yak. Only sites for which the correlation between the observed and imputed data (r2) was >0.9 were retained. To examine potential bias introduced by impute filtering, we compared the SFS before and after filtering. No potential bias was found (Fig. 3c,d). The joint SFS of wild and domestic yaks was used to estimate evolutionary scenario parameters. We used the folded spectrum to minimize potential biases when determining the ancestral allelic states. Alternative models of historical events were fitted to the joint SFS of wild and domestic yak and we allowed only instantaneous population size changes (Supplementary Fig. 6). For each model, we ran the programme 50 times with varying starting points to ensure convergence, and retained the fitting with the highest likelihood. Estimates were obtained from 100,000 simulations per likelihood estimation (-n100,000, -N100,000), 40 Expectation/Conditional Maximization (ECM) cycle (-L40) and 50 runs per data set. The best model was addressed through the maximum value of the likelihoods and Akaike information criterion28. Parametric bootstrap estimates were obtained by parameter estimation based on 100 data sets simulated according to CML estimates in best model (model15) estimation parameters (Supplementary Data 2). The population history and parameters from the best model were used to perform forward simulation and residuals analysis with ∂a∂i (ref. 58) to check the accuracy of the demographic model.
Additional information
Accession codes: The sequencing data for this project have been deposited in the European Nucleotide Archive (EMBL-EBI) under accession code PRJNA285834.
How to cite this article: Qiu, Q. et al. Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat. Commun. 6:10283 doi: 10.1038/ncomms10283 (2015).
Accession codes
References
Diamond, J. Evolution, consequences and future of plant and animal domestication. Nature 418, 700–707 (2002).
Larson, G. et al. Current perspectives and the future of domestication studies. Proc. Natl Acad. Sci. USA 111, 6139–6146 (2014).
Dong, G. et al. Spatial and temporal variety of prehistoric human settlement and its influencing factors in the upper Yellow River valley, Qinghai Province, China. J. Archaeol. Sci. 40, 2538–2546 (2013).
Qi, X. et al. Genetic evidence of paleolithic colonization and neolithic expansion of modern humans on the tibetan plateau. Mol. Biol. Evol. 30, 1761–1778 (2013).
Qiu, J. Who are the Tibetans? Science 347, 708–711 (2015).
Meyer, M. C. et al. Holocene glacier fluctuations and migration of Neolithic yak pastoralists into the high valleys of northwest Bhutan. Quart. Sci. Rev. 28, 1217–1237 (2009).
Zhang, R. C. Yak of China Gansu Scientific and Technical Publishers (1989).
Guo, S. et al. Origin of mitochondrial DNA diversity of domestic yaks. BMC Evol. Biol. 6, 73 (2006).
Wang, Z. F. et al. Phylogeographical analyses of domestic and wild yaks based on mitochondrial DNA: new data and reappraisal. J. Biogeogr. 37, 2332–2344 (2010).
Atlas of Chinese Cultural Relics-Fascicule of the Tibet Autonomous Region Bureau of National Cultural Relics, Cultural Relics Press, Beijing (2010).
Wiener, G., Jianlin, H. & Ruijun, L. The Yak 2nd edn FAO Regional Office for Asia and the Pacific Food and Agriculture Organization of the United Nations, Bangkok, Thailand (2003).
Zhang, M. Q., Xu, X. & Luo, S. J. The genetics of brown coat color and white spotting in domestic yaks (Bos grunniens). Anim. Genet. 45, 652–659 (2014).
Gibbs, R. A. et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324, 528–532 (2009).
Ross-Ibarra, J., Morrell, P. L. & Gaut, B. S. Plant domestication, a unique opportunity to identify the genetic basis of adaptation. Proc. Natl Acad. Sci. USA 104, (Suppl 1): 8641–8648 (2007).
Porto-Neto, L. R. et al. Genome-wide detection of signatures of selection in Korean Hanwoo cattle. Anim. Genet. 45, 180–190 (2014).
Axelsson, E. et al. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 495, 360–364 (2013).
Li, M. et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 45, 1431–1438 (2013).
Ressler, K. J. et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470, 492–497 (2011).
Moreau, M. M. et al. The planar polarity protein Scribble1 is essential for neuronal plasticity and brain function. J. Neurosci. 30, 9738–9752 (2010).
Winberg, M. L. et al. Plexin A is a neuronal semaphorin receptor that controls axon guidance. Cell 95, 903–916 (1998).
Heyne, H. O. et al. Genetic influences on brain gene expression in rats selected for tameness and aggression. Genetics 198, 1277–1290 (2014).
Carneiro, M. et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science 345, 1074–1079 (2014).
Montague, M. J. et al. Comparative analysis of the domestic cat genome reveals genetic signatures underlying feline biology and domestication. Proc. Natl Acad. Sci. USA 111, 17230–17235 (2014).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Zheng, B., Xu, Q. & Shen, Y. The relationship between climate change and Quaternary glacial cycles on the Qinghai–Tibetan Plateau: review and speculation. Quat. Int. 97, 93–101 (2002).
Zhao, S. et al. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat. Genet. 45, 67–71 (2013).
Zhou, X. et al. Whole-genome sequencing of the snub-nosed monkey provides insights into folivory and evolutionary history. Nat. Genet. 46, 1303–1310 (2014).
Excoffier, L. et al. Robust demographic inference from genomic and SNP data. PLoS Genet. 9, e1003905 (2013).
Cai, D. et al. The origins of Chinese domestic cattle as revealed by ancient DNA analysis. J. Archaeol. Sci. 41, 423–434 (2014).
Chen, F. H. et al. Agriculture facilitated permanent human occupation of the Tibetan Plateau after 3600 BP. Science 347, 248–250 (2015).
Zhao, M. et al. Mitochondrial genome evidence reveals successful Late Paleolithic settlement on the Tibetan Plateau. Proc. Natl Acad. Sci. USA 106, 21230–21235 (2009).
Aldenderfer, M. Peopling the Tibetan plateau: insights from archaeology. High Alt. Med. Biol. 12, 141–147 (2011).
Wang, J. et al. Reindeer Ewenki's fading culture. Science 347, 957 (2015).
Green, M. R. & Sambrook, J. Molecular Cloning: A Laboratory Manual Cold Spring Harbor Laboratory Press New York (2012).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010).
Schwartz, S. et al. Human-mouse alignments with BLASTZ. Genome Res. 13, 103–107 (2003).
Qiu, Q. et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 44, 946–949 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Korneliussen, T., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15, 356 (2014).
Skotte, L., Korneliussen, T. S. & Albrechtsen, A. Association testing for next-generation sequencing data using score statistics. Genet. Epidemiol. 36, 430–437 (2012).
Kim, S. Y. et al. Estimation of allele frequency and association mapping using next-generation sequencing data. BMC Bioinformatics 12, 231 (2011).
Kawahara-Miki, R. et al. Whole-genome resequencing shows numerous genes with nonsynonymous SNPs in the Japanese native cattle Kuchinoshima-Ushi. BMC Genomics 12, 103 (2011).
Nielsen, R. et al. SNP calling, genotype calling, and sample allele frequency estimation from New-Generation Sequencing data. PLoS ONE 7, e37558 (2012).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Keane, T. M. et al. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 6, 29 (2006).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Fumagalli, M. et al. Quantifying population genetic differentiation from next-generation sequencing data. Genetics 195, 979–992 (2013).
Fumagalli, M., Vieira, F. G., Linderoth, T. & Nielsen, R. NgsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics 30, 1486–1487 (2014).
Korneliussen, T. S., Moltke, I., Albrechtsen, A. & Nielsen, R. Calculation of Tajima's D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics 14, 289 (2013).
Conesa, A. & Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 619832 (2008).
Hassanin, A. & Ropiquet, A. Molecular phylogeny of the tribe Bovini (Bovidae, Bovinae) and the taxonomic status of the Kouprey, Bos sauveli Urbain 1937. Mol. Phylogenet. Evol. 33, 896–907 (2004).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097 (2007).
Gutenkunst, R. N., Hernandez, R. D., Williamson, S. H. & Bustamante, C. D. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5, e1000695 (2009).
Acknowledgements
The project was supported by the National Natural Science Foundation of China (31322052 and 91331102), the National High-Tech Research and Development Program of China (863 Program, 2013AA102505 3-2), Ministry of Science and Technology of the People's Republic of China (2010DFA34610), International Collaboration 111 Projects of China, Fundamental Research Funds for the Central Universities, 985 and 211 Projects of Lanzhou University.
Author information
Authors and Affiliations
Contributions
J.Li. designed and supervised the project. Q.Q., L.W., K.W., Y.Y., T.M., Z.W., X.Z., Z.N., F.H., R.L. and J.Li. collected and generated the data, and performed the preliminary bioinformatic analyses. Q.Q. and L.W. filtered the data and performed the majority of the population genetic analysis with some contributions from K.W. and Y.Y., Q.Q., L.W. and J.Li. wrote the manuscript with critical input from all the authors, whereas R.A. and J.Lo. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-13, Supplementary Tables 1-6, Supplementary Notes 1-3 and Supplementary References (PDF 1755 kb)
Supplementary Information
Putative regions identified to be under domestication sweeps (XLSX 45 kb)
Supplementary Information
Parameters of models estimated in Fastsimcoal2 (XLSX 14 kb)
Supplementary Information
Summary statistics of ABBA statistics (XLSX 181 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Qiu, Q., Wang, L., Wang, K. et al. Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat Commun 6, 10283 (2015). https://doi.org/10.1038/ncomms10283
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms10283
This article is cited by
-
Whole-genome resequencing of major populations revealed domestication-related genes in yaks
BMC Genomics (2024)
-
Appraisal of some ethnic milk products from minor milch animal species around the world: a review
Journal of Ethnic Foods (2023)
-
Genome-wide identification of SNPs associated with body weight in yak
BMC Genomics (2022)
-
Integration of multi-omics data reveals cis-regulatory variants that are associated with phenotypic differentiation of eastern from western pigs
Genetics Selection Evolution (2022)
-
Analysis of whole-genome re-sequencing data of ducks reveals a diverse demographic history and extensive gene flow between Southeast/South Asian and Chinese populations
Genetics Selection Evolution (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
