
Abstract
Neoproterozoic (1,000–542 Myr ago) Earth experienced profound environmental change, including ‘snowball’ glaciations, oxygenation and the appearance of animals. However, an integrated understanding of these events remains elusive, partly because proxies that track subtle oceanic or atmospheric redox trends are lacking. Here we utilize selenium (Se) isotopes as a tracer of Earth redox conditions. We find temporal trends towards lower δ82/76Se values in shales before and after all Neoproterozoic glaciations, which we interpret as incomplete reduction of Se oxyanions. Trends suggest that deep-ocean Se oxyanion concentrations increased because of progressive atmospheric and deep-ocean oxidation. Immediately after the Marinoan glaciation, higher δ82/76Se values superpose the general decline. This may indicate less oxic conditions with lower availability of oxyanions or increased bioproductivity along continental margins that captured heavy seawater δ82/76Se into buried organics. Overall, increased ocean oxidation and atmospheric O2 extended over at least 100 million years, setting the stage for early animal evolution.
Similar content being viewed by others


Introduction
The history of atmospheric and oceanic redox is strongly interrelated with controls on biological evolution. One of the most significant changes in redox conditions occurred during the late Neoproterozoic Era1, alongside low-latitude ‘Snowball Earth’ glaciations and the emergence of metazoans2. The surface ocean is generally considered to have been oxygenated long before the first Neoproterozoic glaciation, the Sturtian, occurring ∼716 Myr ago1,3,4. However, deeper waters remained anoxic and ferruginous (Fe2+ containing) for much of the Neoproterozoic, with fully oxic conditions becoming widespread (but not global) after the Gaskiers glaciation at ∼580 Myr ago5. Extensive deep-ocean oxygenation in the aftermath of the Gaskiers glaciation may have been a consequence of a rise in atmospheric oxygen driven by an influx of nutrients to the ocean during glacial melting6. Oxygenation may then have permitted the emergence of animal motility leading to predation and bioturbation, as well as biomineralization1. A possible rise in atmospheric oxygen at this time is supported by chromium isotope systematics during the later Neoproterozoic7,8, while an increased range in sulphur isotope compositions of biogenic pyrite has been interpreted to reflect increased atmospheric oxygen penetration into nearshore sediments9,10. Critically, however, proxies that can temporally resolve the progressive oxidation of the late Neoproterozoic and early Cambrian ocean are lacking, as existing proxies often require specific depositional conditions, or are only applicable to tracking more extreme redox changes such as identifying fully anoxic or oxic conditions.
Selenium (Se) isotopes in marine shales are a novel tracer of ocean and atmospheric oxygenation across the Neoproterozoic Oxygenation Event11,12,13,14. Selenium has four oxidation states: +6, +4, 0 and −2, and although the biogeochemical Se cycle shares similarities with that of sulphur, there are distinct differences, including redox transitions. Se oxyanion reduction (both SeVI and SeIV) occurs at significantly higher Eh than the reduction of sulphate to sulphide15, and is closer to that of Fe3+ to Fe2+ (Fig. 1). The combination of multiple redox states, high redox potential and relatively low abundance may also make Se highly sensitive to redox changes16. This sensitivity and associated fractionation mechanisms mean that Se isotopes should have responded to the oxygenation of the deep ocean in the Neoproterozoic when the extent of euxinic and ferruginous water masses contracted significantly5.

In the modern surface ocean, Se exists as selenite (SeIV) or selenate (SeVI) anions (collectively denoted SeOx2−) and dissolved organic Se (denoted Seorg). These species have different behaviour and fates depending on the oxygen levels in the water column. Selenium oxyanions have a short, nutrient-like behaviour and residence time (1,100–9,400 years, although a recent estimate suggests 26,000 years during the Phanerozoic)15,17,18. Organic selenium is common and generally dominates15,19 in surface waters, but is reoxidized at depth in oxic water columns (Fig. 2a) and has very low concentrations below the modern photic zone20. In modern anoxic water columns, in contrast, the remineralization of organic matter is limited, so that Seorg dominates below the chemocline16,20. The sedimentary archive consists of organic and reduced Se, where the former dominates in sediments deposited in anoxic environments16.

(a) (i) Modern cycling of Se in seawater, following Cutter and Bruland47. Underlines indicate relative concentrations in surface and deep water. Reduction of Se and incorporation into phases in sediments can impart negative isotope fractionation. (ii) Possible cycling of Se in Proterozoic seawater when the deep ocean was not oxygenated and had little SeOx2−. Reduction of Se from oxyanions would have been very limited. (b) A simplified diagram showing the main inputs and outputs in the geological selenium cycle16 (analogous to a very similar global scheme developed for isotopes of Mo, which is also a chalcophile element67). The main inputs to the ocean are volcanic and weathering fluxes with δ82/76Se values of δvolc and δweath, respectively22. A key output (highlighted in red) is the largely quantitative removal in isolated euxinic basins, which captures the bulk δ82/76Se value in seawater, δseawater. Another key output, which we sample in our measurements, is removal of Se into sediments on slope or shelf environments in two key components that are mixed in the sediments with Se isotopic composition, δsed11,12,15,22,49. The first form is organically bound selenium (Seorg), which likely captures seawater isotopic composition, δseawater. The second form is selenide (Se2−) or elemental selenium (Se0) produced from the microbial reduction of seawater selenite or selenate (SeOx2−). Because of isotopic fractionation, this second component tends to sequester lighter selenium isotopes. In the above diagram, we ignore small oceanic Se inputs from deep-sea hydrothermal sources and their outputs in the deep sea, because this Se does not mix into the oceans21. Cycling of Se from ocean to atmosphere and back via volatile biogenic organic selenides also does not cause isotope fractionation.
About 85% of the input flux of Se to the oceans (Fig. 2b) arises from rivers, through a combination of organic-bound Se16 and oxidative weathering of continental selenides to SeOx2−; almost all the rest comes from subaerial volcanism via rainfall. Hydrothermal fluid input is small (<1% (ref. 16)), and in any case its Se is probably scavenged by minerals close to the vent21,22. It has also been suggested that hydrothermal fluxes were insignificant in the Archaean and earlier Proterozoic13,16.
Here we reconstruct redox across the second oxygen rise, using marine shale Se isotope ratios. These are novel tracers of oceanic redox conditions, where lighter isotope ratios in shales are generally indicative of a large pool of oxyanions, implying more oxic conditions. There is an overall trend towards lighter isotope ratios from before the Sturtian glaciation to the Ediacaran–Cambrian boundary. Where time periods are covered by data from more than one sample suite, it suggests that the sediment Se isotopes reflect global redox changes. Therefore, we suggest that the global ocean became more oxic from at least the end of the Marinoan glaciation, and that, rather than a sudden redox change, the shift towards deep-ocean oxia was spread over a timescale of ∼100 Myr ago.
Results
Samples
We report Se isotope ratios and Se concentrations for marine shales from seven different geological sections that together span from 770 to 525 Myr ago. Sample suites include the Mackenzie Mountains, Canada (the Twitya and Sheepbed formations), the Yangtze Platform (comprising samples from the Doushantuo, Luichapo and Niutitang formations), Tapley Hill, Australia and finally from the Mineral Fork Formation, and Uinta Mountain and Chuar groups, western USA.
Both formations from Canada are a part of the Windermere Supergroup. The Twitya Formation represents continental slope deposition on a passive margin, where water depths may have reached ∼1 km (ref. 23). In contrast, the Sheepbed Formation was deposited in a deep-water outer shelf setting23. Both Formations contain assemblages of spheromorphic acritarchs, as well as Ediacara-type fossils. The Yangtze Platform, China, comprises several formations according to the nomenclature of ref. 5 (Niutitang, Doushantuo and Liuchanpo). They are thought to represent deposition on the lower continental slope to deep basin of a passive margin setting24,25. The Tapley Hill Formation is from the Adelaide Rift Complex, Australia. It was deposited in a relatively shallow epicontinental basin of about 200–300 km width, with apparent access to the global ocean26.
Four members of the Chuar Group (Grand Canyon, USA) were analysed, including samples from the Walcott Member (742 Myr ago27), the Awatubi Member (∼745 Myr ago upper, 750 Myr ago lower28), the Carbon Canyon Member (∼755 Myr ago) and the Jupiter Member (760 Myr ago28). The group was deposited in an intracratonic extensional basin between ∼740 and 780 Myr ago29. The depositional setting was marine to marginal marine, with access to the open ocean during transgressions, recorded by both sedimentological (wave and tide influenced deposition) and palaeontological evidence30.
Samples from three different Formations of the Uinta Mountain Group (Utah, USA) were also analysed. The group correlates with the Chuar Group, using sedimentological, geochemical, palaeontological and geochronological data31. The Uinta Mountain Group was deposited in an intracratonic extensional basin during the early stages of the breakup of Rodinia31. The quartzose shale of the Red Pine Shale (sample age 740 Myr ago, based on correlation with the Chuar Group) indicates a prodelta marine environment31. The sample from the Moosehorn Lake formation (sample age 765 Myr ago, based on correlation with eastern Uinta Mountain Group formation of Outlaw Trail31) represents a marine setting, which was connected to the open ocean. Finally, the Deadhorse formation (∼760 Myr ago, stratigraphically above the Moosehorn Lake Formation) represents a barrier bar environment, and as such can be considered to be marine, albeit nearshore31.
Finally, a single sample from the Mineral Fork formation was analysed. Zircons in this Formation have been dated at ∼700 Myr ago, and other detrital zircon peaks in correlative successions are dated at 700–680 Myr ago32. Hence, while using the earlier dates of the Sturtian onset from ref. 4 (based partly on Twitya) of ∼717 Myr ago, the maximum depositional age of the Mineral Fork (∼700 Myr ago) suggests a later glacial effect further south32.
Sample chronology is taken from several studies5,31,32. In general, these are based on correlation between absolute ages of individual dated layers. The sample dates between the Sturtian and Marinoan have been adjusted to the new timing of the Sturtian4, by maintaining the relative timing from ref. 5, and condensing the absolute ages.
Shale Se isotope record
Selenium concentrations in our sample shales vary widely (0.02–39.5 μg g−1), with some correlation between [Se] and TOC (total organic carbon; Table 1; Fig. 3). This suggests that some sedimentary Se was buried bound to organic matter, as expected12,33. There is also a generally positive, but poor (r2=0.2) relationship between [Se] and the proportion of highly reactive Fe bound in pyrite, suggesting a weaker effect of substitution for S in pyrite. However, none of these parameters correlate with Se isotope ratios (Fig. 4), showing that δ82/76Se are controlled differently from Se concentrations, which again is anticipated for Se behaviour and may reflect changing reservoir effects over time or between basins12,16.

The co-variation with TOC (total organic carbon), for all samples from all of this study’s different sample locations, suggests that Seorg is an important Se phase in sediments.

The lack of correlations between any of the parameters, as demonstrated by the R2 values and trend lines, shows different controls on Se isotopes from those that control Se concentrations.
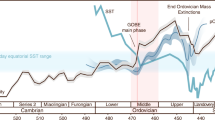
The measured range of our Neoproterozoic samples (−2.35 to +3.63‰, where the Se isotope ratio is reported relative to NIST-3149 (refs 34, 35, 36); Fig. 5; Table 1) is generally similar to the full range of unaltered Phanerozoic and Archaean shale11,12,13,14,16,22,37,38,39, which might be expected if the data reflect a transitional interval from relatively low to higher oxygen levels. Indeed, the data show an overall decrease in δ82/76Se with time, from the highest values in pre-Sturtian sediments, through to the lowest values at the Precambrian–Cambrian boundary (∼550 Myr ago). In detail, the data become isotopically lighter in the run-up to each of the three glaciations, but after the Marinoan glaciation, δ82/76Se is reset at higher values. This return to higher δ82/76Se is not apparent after the Gaskiers glaciation, but an excursion to moderately heavier values occurs during an euxinic interval in the early Cambrian Yangtze Basin. Where samples from two different basins overlap in age (that is, pre-Sturtian, immediately post-Sturtian and between the Marinoan and Gaskiers at 625–584 Myr ago, Fig. 5), we find a remarkable consistency of δ82/76Se values, which may suggest that our data represent global processes rather than local redox changes or diagenetic alteration. It has been proposed that Se isotopes can become highly fractionated during modern oxidative weathering of exposed, very Se-rich (≤26 wt%), outcrops38, where it appears that Se was oxidized and then re-reduced during aqueous transport. However, other studies of Se poorer soils have reported much less fractionation (<0.5‰)40, and the studies of large global data sets also suggest that isotopic effects of late-stage weathering of outcrops are probably not significant12,16. The highly fractionated values from the Se-rich soil38 may thus not be representative of all exposed surface environments. In any case, we present Se data from a combination of cores and outcrops5,31,32, and from different global localities, the combination of which should guard against significant late-stage diagenetic and weathering effects. In addition, for outcrop material, all weathered edges were cut away, and these samples often contain significant pyrite and Fe carbonate minerals suggesting minimal oxidation5, as confirmed by conventional and cathodoluminescence petrographic techniques41. Iron speciation data suggest that most of this study’s samples were ferruginous, albeit with some euxinic conditions at the Precambrian–Cambrian boundary (Table 1)5,42.

The vertical grey bar represents the mantle δ82/76Se43, and the vertical dashed line represents likely pre-industrial modern seawater δ82/76Se12,22. Blue horizontal bars represent glacial intervals. The black histogram shows the range of Precambrian δ82/76Se13, while the grey histogram shows the range of Phanerozoic shale data12,14. Deep-water redox conditions are also shown on the right, based on Fe speciation data for the same sediments. Ferruginous deep-water conditions dominate around the Sturtian and Marinoan glaciations. Post-Gaskiers redox conditions are more variable globally, with evidence for widespread ocean oxygenation, but a continuation of ferruginous conditions in some basins. The diachronous initiation of the Sturtian glaciation is based on the geological evidence (see Sample section).
Discussion
The Se isotopic composition of volcanically derived Se is likely to be similar to that of the mantle (δ82/76Se ∼0–0.25‰ (refs 16, 43)). During oxidative weathering, relatively small fractionations of ∼0–0.4‰ occur40. However, during dissimilatory or abiotic oxyanion reduction in some modern and ancient lakes and rivers, isotopically light Se is sequestered in sediments, driving surface waters heavy by over 3‰ (refs 16, 44). Overall, modern ocean inputs are probably close to ∼0‰ on average, but may have differed in the past. Significant positive values in marine sediments from most of the earlier Precambrian may indicate that fluvial Se was isotopically heavy due to partial reduction during transport16. Marine plankton are thought to assimilate Se without fractionation and the isotopic composition of +0.42±0.22‰ in modern phytoplankton12 suggests that pre-industrial seawater SeOx2− may have had δ82/76Se ∼+0.4‰ (refs 12, 16, 22). This elevated isotope ratio compared with the likely input must be balanced by accumulation of selenium with lower δ82/76Se in average marine sediments12.
Selenium can be preserved in sediments in three different forms. On a global scale, most modern selenium is biologically reduced by assimilation and deposited as Seorg, with little inherent isotope fractionation (<0.9‰)15,16,44,45. Another component of sedimentary Se may be SeIV adsorbed onto Fe-oxyhydroxides. This process also imparts a relatively small isotope fractionation of on average ∼0.15‰ (refs 45, 46). The third component comprises inorganic reduced phases (Se0 and Se−II in sulphide minerals) that form via microbial dissimilatory reduction under suboxic conditions. Given that a significant fraction of the sedimentary Se is associated with organic matter47,48, any potential inorganic Se−II or Se0 phases that formed by oxyanion reduction and may carry more negative δ82/76Se values become diluted16. This may explain why natural bulk sediments generally show much smaller fractionations relative to Se sources to the ocean than what is observed in laboratory experiments16.
Isotopic fractionation of selenium species in sediments is related to the redox state of the overlying water. Both experimental11 and ab initio calculations49 have reported fractionation of Δ82/76Seoxyanions−SeO ≤−21‰, although natural fractionation is considerably smaller12,16. In any case, ab initio equilibrium calculations likely do not reflect the largely kinetic fractionation likely occurring in natural systems47,50. The additional reduction of Se0 to selenide is not associated with significant isotope fractionation11. Thus, where dissimilatory reduction of SeOx2− occurs, it sequesters lighter isotopes into sediments. If the site of the reduction (for example, suboxic sediments or oxygen-minimum zones (OMZs) in the water column) is linked to a large Se oxyanion reservoir, as, for example, the modern oxic ocean, then SeOx2− reduction may be non-quantitative, such that sedimentary reduced phases preserve large negative fractionations14,15,39. Therefore, one would expect that an increase in the size of the dissolved SeOx2− reservoir correlates with larger and more negative fractionations in locally suboxic environments. Selenium isotope compilations from across geological time statistically support this relationship, with mean isotope ratios heaviest in the mid-Archaean before the onset of oxidative weathering around ∼2.7 Gyr ago (δ82/76Se=+0.93±0.41‰, 1 s.d.; Fig. 6a), intermediate in the early to middle Proterozoic (+0.68±0.83‰, up to ∼1.1 Gyr ago), and lightest in the Phanerozoic (−0.30±0.89‰)16. Shorter-term trends in sedimentary Se isotope ratios confirming the same patterns have been reported from the latest Archaean13, the Permo–Triassic (P-Tr) boundary39, Mesozoic Oceanic Anoxic Events (OAEs; although these appear to exhibit basinal gradations12,16) and glacial–interglacial redox variations in the Cariaco Basin22.

(a) Published data (open symbols)16 compared with data from this study (closed squares) (n=463). The horizontal lines represent the mean from each time period16, demonstrating the overall trend towards isotopically lighter sediments after each major oceanic oxidation stage. (b) A focus of the same data compilation to the time period of this study. Here the black lines show the data mean from 1,500 Myr ago to the Sturtian (+0.92±0.76‰), and from the Gaskiers to the present (−0.32±0.85‰).
The interpretation of a link between redox trends and Se isotopic composition may be complicated by temporal variations in the compositions of inputs to the oceans (dominantly riverine fluxes), and by local redox changes in restricted basins or marginal seas that are not representative of the global ocean. The former implies that seawater δ82/76Se has changed at key intervals in Earth history, as also suggested for Mo isotopes51,52. The latter is particularly important for Se due to its short marine residence time and its high sensitivity to changes in biological productivity39. Locally enhanced productivity may increase the relative proportion of Seorg, which would lead to a smaller net fractionation of bulk sediments relative to seawater, because the dissolved SeOx2− reservoir becomes locally depleted and because Seorg dilutes any fractionation carried by inorganic reduced phases. Hence, global, rather than local, redox conditions can only be inferred with confidence if similar trends and values occur in multiple different settings of similar age. Therefore, as with all tracers of marine redox conditions (for example, Mo isotopes53), Se isotopes provide information on local water column and water-sediment interface conditions, with an additional control from global seawater isotope ratios.
Published modern (<500 kyr old) data clearly show that sediments deposited in the oxic open ocean, or under well-connected OMZs, have isotopically lighter Se than those deposited in anoxic, restricted basin conditions16,22. The lighter values probably reflect the large SeOx2− reservoir in oxic seawater, which allows for incomplete dissimilatory reduction under locally suboxic conditions, such as during early diagenesis in sediments or in OMZs. In contrast, sediments deposited under anoxic or euxinic conditions incur little to no isotopic fractionation, likely due to a combination of more quantitative SeOx2− reduction, a higher proportion of Seorg, and a local increase in the isotopic composition of dissolved SeOx2− due to Rayleigh distillation16. In ferruginous systems, preferential removal of isotopically light Se by abiotic reduction with Fe(II) may play a role, with experimental results suggesting large fractionations, although it is unknown whether these experiments reflect the degree of fractionation in a natural state11,14,16,46,54. It has been suggested that light Se isotope ratios reported from the early Cambrian Yangtze platform may be due to this process14, but in this case the ferruginous reduction site was likely linked to a large Se oxyanion reservoir such as the global ocean or a deep oxic surface layer16,46. Overall, over geologic time, marine sediments have become isotopically lighter, in response to the overall oxidation of the ocean-atmosphere system (Fig. 6)16. The trend towards more negative values in our Neoproterozoic δ82/76Se data (Fig. 5) is thus most plausibly interpreted as deep-water oxygenation and an increase in the marine SeOx2− reservoir towards the Precambrian–Cambrian boundary. Excursions from other time periods (latest Archaean13, P–Tr boundary39, OAEs12,16 and glacial–interglacial redox variations22) have been interpreted in a similar manner, lending support to our Neoproterozoic interpretation.
One key observation is that δ82/76Se values before the Sturtian and immediately after the Marinoan are isotopically heavier than both the mantle and average Precambrian values (Fig. 5). The highest Precambrian δ82/76Se value published before this study is +3.04‰ from ∼2.3 Gyr ago16, compared with our highest values of +3.63‰ at 0.765 Gyr ago, and positive values are generally observed in the Archaean16. Highly positive values are also reported from the first ‘whiff of oxygen’13 and during a local euxinic event shortly before the Permo–Triassic mass extinction39.
Several possibilities can be proposed for driving sediment δ82/76Se higher than crust or mantle values in the Precambrian. The first is that Precambrian rivers became isotopically heavy. Early rivers could have contained a large fraction of organically complexed Se, due to land-based microbial life. In this case, increasing atmospheric oxygen could have increased the SeOx2−/Seorg ratio, generating more reduction of SeOx2− in rivers, driving the residue to higher δ82/76Se values16. This scenario is reasonable given that late Archaean lake and river sediments are isotopically lighter than marine sediments from the same time, indicating heavier continental surface waters16, assuming a similar fractionation mechanism in fresh and seawater. This phenomenon has also been reported from modern lakes with changing redox44. As during the late Archaean ‘whiff of oxygen’13, the pre-Sturtian δ82/76Se peak in our data set may thus reflect an atmospheric rise in pO2 above background levels without significant seawater oxygenation. A second possibility is that a Se isotope gradient with ocean depth could have formed, where continuous removal of light Se into sediments in shallower oxic waters would enrich offshore waters in heavy isotopes, also driving sediments downslope to isotopically heavier compositions. Such behaviour has been proposed to explain Archaean Se data13, as well as suggested for molybdenum isotopes13,53. However, it is only likely to apply here if the intracratonic basin represented by the Uinta Mountain and Chuar data had a universally deep chemocline everywhere in the basin. Hence, the isotopically heavy data might indicate that rivers became isotopically heavier at these times, likely due to relatively sudden increases in atmospheric O2 during the run-up to oceanic oxygenation. In fact, the most positive values around 770 Myr ago approximately coincide with a large perturbation in Cr isotopes, which has been interpreted as evidence of atmospheric oxygenation before the Sturtian glaciation7. Given that during the Marinoan CO2 levels appear to have increased55,56,57, and therefore O2 levels decreased, there could have been another O2 rise soon after the Marinoan, which may again have led to isotopically heavy Se in river waters. The increasing availability of oxygen is also consistent with high proportions of organic carbon burial in interglacial times58. Exceptionally high δ82/76Se is not observed after the Sturtian, but this may be because our samples are slightly older relative to deglaciation, compared with the post-Marinoan samples. A third possibility for high δ82/76Se values is high organic carbon burial. Enhanced biological productivity along continental margins could also have directly contributed to the observed heavy δ82/76Se after the Marinoan glaciations, because such conditions may have locally enhanced the proportion of Seorg in sediments.
Changing riverine Se isotope ratios are unlikely to have caused the trend towards isotopically light values observed in our samples, because there is no plausible mechanism that would drive river waters isotopically light. The few riverine sediments from the geologic past that have been measured have significantly lighter δ82/76Se than the crustal value16, implying isotopically heavy waters. Hence the negative fractionations in marine sediments are likely the result of in situ SeOx2− reduction, either within sedimentary pore waters or in suboxic bottom waters. Importantly, the preservation of light δ82/76Se values relative to the crust implies that reduction was not quantitative. One possible explanation is a massive drop in productivity, such as observed during the P–Tr extinction39, which would lead to a decline in Seorg deposition. Although carbon burial may indeed have declined after its initial post-glacial boost58, there is no overall correlation between Se and C isotopes (Fig. 7). Therefore, the trend towards lighter values is probably a reflection of an increasing SeOx2− reservoir in the Neoproterozoic ocean, that is, an increase in Se supply rather than a decrease in demand. The fact that multiple basins of similar age show similar patterns suggests that dissolved SeOx2− concentrations increased globally, which is consistent with widespread oxygenation of the deep ocean.

Se isotopes suggest that the oxidation of the oceans took at least 100 Myr ago. The lack of correlation with δ13C suggests that the processes that control carbon (burial and oxidation of organic matter and carbonate dissolution/formation)68 appear not to be controlling Se isotopes, meaning that Se is not controlled by organic matter. The timing of the Sturtian has been altered according to ref. 4 (Sturtian is not shown as diachronous, as opposed to Fig. 5, for simplicity’s sake).
A key complication in interpreting sedimentary Se isotopes (or indeed any redox proxy) is the influence of local redox changes in the water column, relative to global oceanic oxidation changes. Localized changes in redox (for example, ‘euxinic wedges’) are thought to have occurred towards the end of the Cryogenian, especially at intermediate depths5,59,60. Such local changes would be assumed to complement or override Se isotope variations caused by global ocean changes, as, for example, also shown for redox tracers such as Mo and Cr isotopes7,8,53. In addition, the closer-to-quantitative capture of Se in more reduced settings would likely decrease the Se residence time before oxygenation12,16, enhancing local effects. The clearest method for attempting to determine a local versus global Se isotope signal is to compare sample suites from different global locations. Thus, similar δ82/76Se values between the data from Chuar and Uinta Mountain groups between ∼770 and 742 Myr ago suggest that, at least in the intracratonic basin associated with these groups, Se isotope ratios rapidly decreased. Immediately after the Sturtian (662–659 Myr ago), our Twitya and Tapley Hill samples give similar δ82/76Se values, and at times between the Marinoan and Gaskiers (∼625–584), similar isotope ratios are seen from the Sheepbed and Yangtze sample suites. Given that these suites are from different continents and different ocean depths, this suggests that Se isotope ratios between the Sturtian and Gaskiers generally represent global ocean phenomena (except, potentially, high δ82/76Se in the Sheepbed samples immediately after the Marinoan). Combined, these samples nevertheless suggest that global ocean sediments were becoming isotopically lighter during the course of the end-Cryogenian and Ediacaran.
Data after the Gaskiers glaciation only exist from the Yangtze Platform, and, therefore, there is nothing to prove that trends recorded here are more than local redox changes. However, the large δ82/76Se variation recorded from the Yangtze (from −2.3‰ to +0.4‰, followed by a return to negative values) provide a natural experiment that supports our interpretation of a redox control on δ82/76Se. At ∼540 Myr ago, Fe speciation suggests a localized, transient, ∼10 Myr interval of euxinia5. Once euxinia starts, δ82/76Se values increase substantially, and approach ∼0‰, reflecting near-quantitative removal of Se from seawater under euxinic conditions, in the form of sulphides and Seorg (refs 12, 13, 61). Sections from elsewhere in the Yangtze Basin display similarly high δ82/76Se values14 (Fig. 5). Then, once Fe speciation indicates a cessation of euxinia, δ82/76Se decreases again. Thus, sediments deposited under more reducing conditions have significantly higher δ82/76Se values than those deposited under more oxic conditions, as also implied by rapid12,13,39 and long-term16 oceanic trends.
Selenium’s redox transitions result in Se isotopes being sensitive to redox conditions between those of S−II–SIV and FeII–FeIII. Hence, our data imply that at ∼770 Myr ago, coastal to intermediate depth (the depositional environment of the Chuar and Uinta Mt. groups) Eh was below the SeIV–Se0 transition, with reducing conditions widespread on shallow shelves, coupled to likely changes in the riverine isotope ratio, in response to atmospheric oxygenation. Afterwards, temporal trends in δ82/76Se imply that the oceans became progressively oxidized, as evidenced by deep marine depositional environments from different global locations showing increasingly lower δ82/76Se values. Global seawater reached a minimum around the Gaskiers glaciation (as shown by low values in both the Sheepbed and Yangtze), and potentially a lower minimum at the Precambrian–Cambrian boundary, although local effects cannot be distinguished here, because only one location was sampled from this time period. It is interesting to note that other samples of roughly similar age (also from the Yangtze Platform14) show even lighter Se isotope ratios (Fig. 6).
Overall, the Neoproterozoic data from this study fit well into the trends of Se isotope data over geologic time16 (Fig. 6). Samples before the Sturtian and after the Marinoan glaciations are isotopically heavier than the next youngest samples from ∼400 to 700 Myr ago earlier16. As discussed above, heavy values before the Sturtian could be a function of local conditions during intracratonic basinal deposition, where riverine influence on shallow waters would be relatively greater, and may not reflect wider global conditions. Heavy values after the Marinoan are also only reflected by one sample suite, but could also reflect changing continental weathering conditions during recovery from the ‘snowball’ glaciation.
Hence, our data appear to capture an important transitional interval from high δ82/76Se values typical of the Archaean and early Proterozoic12,13,16 to the lighter values that characterise the Phanerozoic12,14,16,22,35,38,39 (Figs 5 and 6). This shift is consistent with oxygenation of the deep ocean during this time period, and in particular, suggests that the shift between fully ferruginous and fully oxic deep oceans took considerable time. The possibility that the trend towards lower δ82/76Se was caused by a relaxation in organic burial (and hence less heavy Seorg burial) is unlikely because there is no correlation between δ82/76Se and δ13C (Fig. 7).
The selenium isotope data imply that oxygenation was a protracted process. Iron speciation indicates that fully oxic deep waters only formed after the Gaskiers glaciation5. The Se isotope data suggest that this oxidation was spread over time, likely starting after the Marinoan, and taking around 100 Myr to pass through ferruginous and intermediate redox conditions (for example, manganous) to reach full deep-ocean oxia. Selenium’s low abundance, multiple redox states and high redox potential may make Se isotopes a more sensitive redox proxy than tracers such as sulphur or molybdenum isotopes16, or ‘on-off’ tracers such as Fe speciation. Hence, these data provide insight into the small degrees of redox change that led, perhaps in fits and starts, to full oceanic oxygenation. A Cryogenian–Ediacaran increase in oxygenation agrees with other redox-sensitive elements10,62,63 and biomarker evidence64. Thus, the significance of the Se isotope record is not only that it adds to growing evidence that the late Proterozoic and Cambrian ocean and atmosphere reached a progressively more oxic state, coinciding with the diversification of animal life, but also that the process of oxidation was protracted, and not ultimately triggered by the Gaskiers deglaciation, as other data suggest5.
In summary, this study has determined Se isotope ratios from shales deposited across the Neoproterozoic Oxygenation Event, from 770 to 525 Myr ago. Selenium isotopes are sensitive to changing oceanic redox conditions, both global and local, where lighter isotope ratios in sediments imply deposition from a water body that was linked to a large, oxic SeOx2− reservoir. This hypothesis is supported by studies of other geologic time periods, as well as by local redox variations recorded by our post-Gaskiers samples. Overall, we find a trend of decreasing sediment δ82/76Se with time, and where time periods are covered by data from more than one sample suite, it suggests that the sediment Se isotopes reflect global redox changes. Hence, the data suggest that the global ocean became more oxic from at least the end of the Marinoan glaciation, and that, rather than a sudden redox change, the shift towards deep-ocean oxia was spread over a timescale of ∼100 Myr.
Methods
Analytical methods
The analytical methods used here for Se isotopes have been reported elsewhere35,36, but, briefly, two separate methods were used, where both report relative to NIST SRM-3149 (ref. 34). Samples younger than the Sturtian glaciation (∼670 Myr ago) were analysed by a double-spike inversion method, where international rock standard results were double-checked using different spikes and inversions, as well as doping and inverse modelling experiments35. The chemistry and analyses were performed at the Bristol Isotope Group, UK, using a Thermo Neptune MC-ICP-MS. Data from before the Sturtian were obtained using a sample-standard bracketing technique, with analyses performed on a Nu Instruments MC-ICP-MS at the University of Washington, USA36. To compare the techniques, the USGS shale standard SGR-1 was analysed by both methods. The former method yields δ82/76Se=+0.25±0.17‰ (n=16), and the latter +0.05±0.18‰ (n=9), meaning that values are identical within analytical uncertainty35,36.
Additional information
How to cite this article: Pogge von Strandmann, P. A. E. et al. Selenium isotope evidence for progressive oxidation of the Neoproterozoic biosphere. Nat. Commun. 6:10157 doi: 10.1038/ncomms10157 (2015).
References
Shields-Zhou, G. & Och, L. The case for a Neoproterozoic Oxygenation Event: Geochemical evidence and biological consequences. Geol. Soc. Am. Today 21, 4–11 (2011).
Narbonne, G. M. The ediacarabiota: Neoproterozoic origin of animals and their ecosystems. Annu. Rev. Earth Planet. Sci. 33, 421–442 (2005).
Macdonald, F. A. et al. Calibrating the Cryogenian. Science 327, 1241–1243 (2010).
Rooney, A. D. et al. Re-Os geochronology and coupled Os-Sr isotope constraints on the Sturtian snowball Earth. Proc. Natl Acad. Sci. USA 111, 51–56 (2014).
Canfield, D. E. et al. Ferruginous conditions dominated later neoproterozoic deep-water chemistry. Science 321, 949–952 (2008).
Canfield, D. E., Poulton, S. W. & Narbonne, G. M. Late-Neoproterozoic deep-ocean oxygenation and the rise of animal life. Science 315, 92–95 (2007).
Planavksy, N. J. et al. Low mid-Proterozoic atmospheric oxygen levels and the delayed rise of animals. Science 346, 635–638 (2014).
Frei, R., Gaucher, C., Poulton, S. W. & Canfield, D. E. Fluctuations in Precambrian atmospheric oxygenation recorded by chromium isotopes. Nature 461, 250–253 (2009).
Canfield, D. E. The Early History of Atmospheric Oxygen. Annu. Rev. Earth Planet. Sci. 33, 1–36 (2005).
Turner, E. C. & Bekker, A. Thick sulfate evaporite accumulations marking a mid-Neoproterozoic oxygenation event (Ten Stone Formation, Northwest Territories, Canada). Geol. Soc. Am. Bull. B31268.1 (2015).
Johnson, T. M. & Bullen, T. D. in Geochimistry of Non-Traditional Stable Isotopes eds Johnson C. M., Beard B. L., Albarede F. 289–317Geochemical Society (2004).
Mitchell, K. et al. Selenium as paleo-oceanographic proxy: a first assessment. Geochim. Cosmochim. Acta 89, 302–317 (2012).
Stüeken, E. E., Buick, R. & Anbar, A. D. Selenium isotopes support free O2 in the latest Archean. Geology 43, 259–262 (2015).
Wen, H. J. et al. Selenium isotopes trace anoxic and ferruginous seawater conditions in the Early Cambrian. Chem. Geol. 390, 164–172 (2014).
Fernandez-Martinez, A. & Charlet, L. Selenium environmental cycling and bioavailability: a structural chemist point of view. Rev. Environ. Sci. Biotechnol. 8, 81–110 (2009).
Stüeken, E. E. et al. The evolution of the global selenium cycle: secular trends in Se isotopes and abundances. Geochim. Cosmochim. Acta 162, 109–125 (2015).
Mackenzie, F. T., Lantzy, R. J. & Paterson, V. Global trace metal cycles and predictions. Math. Geol. 11, 99–142 (1979).
Large, R. R. et al. Cycles of nutrient trace elements in the Phanerozoic ocean. Gondwana Res. 28, 1282–1293 (2015).
Cutter, G. A. & Cutter, L. S. Sources and cycling of selenium in the western and equatorial Atlantic Ocean. Deep Sea Res. II 48, 2917–2931 (2001).
Cutter, G. A. Selenium in reducing waters. Science 217, 829–831 (1982).
Layton-Matthews, D. et al. Multiple sources of selenium in ancient seafloor hydrothermal systems: Compositional and Se, S, and Pb isotopic evidence from volcanic-hosted and volcanic-sediment-hosted massive sulfide deposits of the Finlayson Lake District, Yukon, Canada. Geochim. Cosmochim. Acta 117, 313–331 (2013).
Shore, A. J. T. Selenium geochemistry and isotopic composition of sediments from the Cariaco Basin and the Bermuda Rise: a comparison between a restricted basin and the open ocean over the last 500 ka (PhD thesis Univ. Leicester (2010).
Narbonne, G. M. & Aitken, J. D. Neoproterozoic of the mackenzie mountains, Northwestern Canada. Precambrian Res. 73, 101–121 (1995).
Goldberg, T., Poulton, S. W. & Strauss, H. Sulphur and oxygen isotope signatures of late Neoproterozoic to early Cambrian sulphate, Yangtze Platform, China: Diagenetic constraints and seawater evolution. Precambrian Res. 137, 223–241 (2005).
Goldberg, T., Strauss, H., Guo, Q. & Liu, C. Reconstructing marine redox conditions for the early Cambrian Yangtze Platform: evidence from biogenic sulphur and organic carbon isotopes. Palaeogeogr. Palaeoclimatol. Palaeoecol. 254, 175–193 (2007).
Coats, R. P., Forbes, B. G. & Preiss, W. V. The Adelaide Geosyncline. Late Proterozoic Stratigraphy, Sedimentation, Palaeontology and Tectonics Ministry of Mines and Energy (1987).
Karlstrom, K. E. et al. Chuar Group of the Grand Canyon: record of breakup of Rodinia, associated change in the global carbon cycle, and ecosystem expansion by 740Ma. Geology 28, 619–622 (2000).
Williams, M. et al. Dating sedimentary sequences: in situ U/Th-Pb microprobe dating of early diagenetic monazite and Ar-Ar dating of marcasite nodules: case study from Neoproterozoic black shales in the southwestern U.S. Geol. Soc. Am. Abst. Program. 35, 595 (2003).
Dehler, C. M., Karlstrom, K. E., Gehrels, G. E., Timmons, J. M. & Crossey, L. C. Stratigraphic revision, provenance, and new age constraints of the Nankoweap Formation and Chuar Group, Grand Canyon Supergroup, Grand Canyon, Arizona. Geol. Soc. Am. Abst. Program. 44, 82 (2012).
Dehler, C. M. et al. Neoproterozoic Chuar Group (∼800-742Ma), Grand Canyon: A record of cyclic marine deposition during global climatic and tectonic transitions Vol. 141–142, (Sedimentary Geology (2001).
Dehler, C. M., Fanning, C. M., Link, P. K., Kingsbury, E. M. & Rybczynski, D. Maximum depositional age and provenance of the Uinta Mountain Group and Big Cottonwood Formation, northern Utah: paleogeography of rifting western Laurentia. Geol. Soc. Am. Bull. 122, 1686–1699 (2010).
Yonkee, W. A. et al. Tectono-stratigraphic framework of Neoproterozoic to Cambrian strata, west-central U.S.: protracted rifting, glaciation, and evolution of the North American Cordilleran margin. Earth Sci. Rev. 136, 59–95 (2014).
Gustafsson, J. P. & Johnsson, L. The Association between Selenium and Humic Substances in Forested Ecosystems - Laboratory Evidence. Appl. Organometal. Chem. 8, 141–147 (1994).
Carignan, J. & Wen, H. J. Scaling NIST SRM 3149 for Se isotope analysis and isotopic variations of natural samples. Chem. Geol. 242, 347–350 (2007).
Pogge von Strandmann, P. A. E., Coath, C. D., Catling, D. C., Poulton, S. W. & Elliott, T. Analysis of mass dependent and mass independent selenium isotope variability in black shales. J. Anal. Atom. Spectrom. 29, 1648–1659 (2014).
Stüeken, E. E., Foriel, J., Nelson, B. K., Buick, R. & Catling, D. C. Selenium isotope analysis of organic-rich shales: advances in sample preparation and isobaric interference correction. J. Anal. Atom. Spectrom. 28, 1734–1749 (2013).
Wen, H. J. & Carignan, J. Selenium isotopes trace the source and redox processes in the black shale-hosted Se-rich deposits in China. Geochim. Cosmochim. Acta 75, 1411–1427 (2011).
Zhu, J. M., Johnson, T. M., Clark, S. K., Zhu, X. K. & Wang, X. L. Selenium redox cycling during weathering of Se-rich shales: A selenium isotope study. Geochim. Cosmochim. Acta 126, 228–249 (2014).
Stüeken, E. E., Foriel, J., Buick, R. & Schoepfer, S. D. Selenium isotope ratios, redox changes and biological productivity across the end-Permian mass extinction. Chem. Geol. 410, 28–39 (2015).
Schilling, K., Johnson, T. M. & Wilcke, W. Selenium Partitioning and Stable Isotope Ratios in Urban Topsoils. Soil Sci. Soc. Am. J. 75, 1354–1364 (2011).
Dehler, C. M. et al. High-resolution δ13C stratigraphy of the Chuar Group (ca. 770 –742Ma), Grand Canyon: Implications for mid-Neoproterozoic climate change. Geol. Soc. Am. Bull. 117, 32–45 (2005).
Johnston, D. T. et al. An emerging picture of Neoproterozoic ocean chemistry: insight from the Chuar Group, Grand Canyon, USA. Earth Planet. Sci. Lett. 290, 64–73 (2010).
Rouxel, O., Fouquet, Y. & Ludden, J. N. Subsurface processes at the Lucky Strike hydrothermal field, mid-Atlantic Ridge: evidence from sulfur, selenium, and iron isotopes. Geochim. Cosmochim. Acta 68, 2295–2311 (2004).
Clark, S. K. & Johnson, T. M. Selenium stable isotope investigation into selenium biogeochemical cycling in a lacustrine environment: Sweitzer Lake, Colorado. J. Environ. Qual. 39, 2200–2210 (2010).
Johnson, T. M., Herbel, M. J., Bullen, T. D. & Zawislanski, P. T. Selenium isotope ratios as indicators of selenium sources and oxyanion reduction. Geochim. Cosmochim. Acta 63, 2775–2783 (1999).
Mitchell, K., Couture, R.-M., Johnson, T. M., Mason, P. R. D. & Van Cappellen, P. Selenium sorption and isotope fractionation: iron(III) oxides versus iron(II) sulfides. Chem. Geol. 342, 21–28 (2013).
Cutter, G. A. & Bruland, K. W. The marine biogeochemistry of selenium: a re-evaluation. Limnol. Oceanogr. 29, 1179–1192 (1984).
Baines, S. B., Fisher, N. S., Doblin, M. A. & Cutter, G. A. Uptake of dissolved organic selenides by marine phytoplankton. Limnol. Oceanogr. 46, 1936–1944 (2001).
Li, X. F. & Liu, Y. Equilibrium Se isotope fractionation parameters: a first-principles study. Earth Planet. Sci. Lett. 304, 113–120 (2011).
Kulp, T. R. & Pratt, L. M. Speciation and weathering of selenium in Upper Cretaceous chalk and shale from South Dakota and Wyoming, USA. Geochim. Cosmochim. Acta 68, 3687–3701 (2004).
Archer, C. & Vance, D. The isotopic signature of the global riverine molybdenum flux and anoxia in the ancient oceans. Nat. Geosci. 1, 597–600 (2008).
Pearce, C. R., Burton, K. W., Pogge von Strandmann, P. A. E., James, R. H. & Gislason, S. R. Molybdenum isotope behaviour accompanying continental weathering and riverine transport in a basaltic terrain. Earth Planet. Sci. Lett. 295, 104–114 (2010).
Poulson, R. L., Siebert, C., McManus, J. & Berelson, W. M. Authigenic molybdenum isotope signatures in marine sediments. Geology 34, 617–620 (2006).
Charlet, L. et al. Electron transfer at the mineral/water interface: selenium reduction by ferrous iron sorbed on clay. Geochim. Cosmochim. Acta 71, 5731–5749 (2007).
Hoffman, P. F., Kaufman, A. J., Halverson, G. P. & Schrag, D. P. A Neoproterozoic snowball Earth. Science 281, 1342–1346 (1998).
Kasemann, S. A., Hawkesworth, C., Prave, A. R., Fallick, A. E. & Pearson, P. N. Boron and calcium isotope composition in Neoproterozoic carbonate rocks from Namibia: evidence for extreme environmental change. Earth Planet. Sci. Lett. 231, 73–86 (2005).
Kasemann, S. A. et al. Calcium and magnesium isotopes elucidate changes in continental weathering following a Cryogenian glaciation. Earth Planet. Sci. Lett. 396, 66–77 (2014).
Och, L. M. & Shields-Zhou, G. A. The Neoproterozoic oxygenation event: Environmental perturbations and biogeochemical cycling. Earth Sci. Rev. 110, 26–57 (2012).
Lenton, T. M., Boyle, R. A., Poulton, S. W., Shields-Zhou, G. A. & Butterfield, N. J. Co-evolution of eukaryotes and ocean oxygenation in the Neoproterozoic era. Nat. Geosci 7, 257–265 doi:10.1038/ngeo2108 (2014).
Poulton, S. W. & Canfield, D. E. Ferruginous conditions: a dominant feature of the ocean through Earth's history. Elements 7, 107–112 (2011).
Pettine, M., Gennari, F., Campanella, L., Casentini, B. & Marani, D. The reduction of selenium(IV) by hydrogen sulfide in aqueous solutions. Geochim. Cosmochim. Acta 83, 37–47 (2012).
Sahoo, S. K. et al. Ocean oxygenation in the wake of the Marinoan glaciation. Nature 489, 546–549 (2012).
Gallagher, M., Turner, E. C. & Kamber, B. S. In situ trace metal analysis of Neoarchaean – Ordovician shallow-marine microbial-carbonate-hosted pyrites. Geobiology 13, 316–339 (2015).
Love, G. D. et al. Fossil steroids record the appearance of Demospongiae during the Cryogenian period. Nature 457, 718–722 (2009).
Takeno, N. Atlas of Eh-pH diagrams–Intercomparison of thermodynamic databases (2005).
Ohnemueller, F., Prave, A. R., Fallick, A. E. & Kasemann, S. A. Ocean acidification in the aftermath of the Marinoan glaciation. Geology 42, 1103–1106 (2014).
Anbar, A. D. & Rouxel, O. Metal stable isotopes in paleoceanography. Annu. Rev. Earth Planet. Sci. 35, 717–746 (2007).
Halverson, G. P., Hoffman, P. F., Schrag, D. P., Maloof, A. C. & Rice, A. H. N. Toward a Neoproterozoic composite carbon-isotope record. Bull. Geol. Soc. Am. 117, 1181–1207 (2005).
Acknowledgements
We thank Chris Coath for assistance during method development. Graham Shields is thanked for discussions on C isotopes. This study was funded by NERC grant NE/F016832/1. P.A.E.P.v.S. is supported by NERC fellowship NE/I020571/2.
Author information
Authors and Affiliations
Contributions
P.A.E.P.v.S. carried out most of the analyses, interpreted the data and wrote the manuscript, with contributions from all co-authors. E.E.S. supplied some of the Se isotope data, S.W.P., C.M.D. and D.E.C. provided samples and geochemical context. D.C.C. and T.E. wrote the original NERC research proposal, and T.E. helped in method development.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pogge von Strandmann, P., Stüeken, E., Elliott, T. et al. Selenium isotope evidence for progressive oxidation of the Neoproterozoic biosphere. Nat Commun 6, 10157 (2015). https://doi.org/10.1038/ncomms10157
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms10157
This article is cited by
-
Early Cambrian highly metalliferous black shale in South China: Cu and Zn isotopes and a short review of other non-traditional stable isotopes
Mineralium Deposita (2022)
-
Origin of the early Ediacaran chert from Infrakrol Formation in Krol Belt, Lesser Himalaya, India
Journal of Sedimentary Environments (2022)
-
A Review of the Neoproterozoic Global Glaciations and a Biotic Cause of Them
Earth Systems and Environment (2021)
-
Integrated records of environmental change and evolution challenge the Cambrian Explosion
Nature Ecology & Evolution (2019)
-
A tectonically driven Ediacaran oxygenation event
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
