
Abstract
Targeted delivery of antigens to dendritic cells (DCs) is a promising vaccination strategy. However, to ensure immunity, the approach depends on coadministration of an adjuvant. Here we ask whether targeting of both adjuvant and antigen to DCs is sufficient to induce immunity. Using a protein ligation method, we develop a general approach for linking the immune stimulant, poly dA:dT (pdA:dT), to a monoclonal antibody (mAb) specific for DEC205 (DEC). We show that DEC-specific mAbs deliver pdA:dT to DCs for the efficient production of type I interferon in human monocyte-derived DCs and in mice. Notably, adaptive T-cell immunity is elicited when mAbs specific for DEC–pdA:dT are used as the activation stimuli and are administered together with a DC-targeted antigen. Collectively, our studies indicate that DCs can integrate innate and adaptive immunity in vivo and suggest that dual delivery of antigen and adjuvant to DCs might be an efficient approach to vaccine development.
Similar content being viewed by others


Main
Vaccination is the most valuable approach to induce protective immunity against infectious diseases. Defined proteins or antigens are increasingly used in vaccines, which have improved safety profiles and ease of production relative to intact microbes or microbial vectors1. However, defined antigens are poorly immunogenic for T-cell immunity when administered alone, inducing unresponsiveness or tolerance. Therefore, they must be administered with an adjuvant that triggers immune stimulation and activation2.
Adjuvants are microbial-derived agents or synthetic microbial mimics that activate innate immunity after interacting with pattern recognition receptors (PRRs). Double-stranded RNA (dsRNA) and unmethylated CpG DNA are adjuvants recognized by a group of integral membrane PRRs called Toll-like receptors (TLRs), that is, TLR3 and 9, respectively (reviewed in ref. 3). Cytosolic dsRNA is also an immune activator sensed by the retinoic acid–inducible gene (RIG) I-like receptor family, which include RIG-I and MDA5 (ref. 4). In addition, cytosolic accumulation of double-stranded DNA (dsDNA) is a potent adjuvant that induces the activation of innate immune signaling pathways5,6,7. Recent reports show that detection of cytosolic dsDNA occurs by multiple mechanisms but is independent of TLRs8,9,10. One pathway of activation by cytoplasmatic DNA involves the initial transcription of dsDNA into dsRNA by RNA polymerase III, following the activation of cytoplasmic RIG-I11,12. This Pol III–RIG-I signaling pathway occurs in both human and mouse cells but is redundant in the latter with a still-undefined dsDNA-sensing mechanism, which seems to be independent of RNA polymerase III and RIG-I11. Irrespective of the recognition pathway, several studies indicate that cytosolic dsDNA induces type I interferon (IFN) production, which exerts antimicrobial effects by switching on the transcription of proinflammatory cytokines and antipathogen genes5,6.
DCs are antigen-presenting cells specialized for the initiation and regulation of immune responses13. DCs express an array of PRRs, including TLR and the RIG-I–like and C-type lectin receptors, which enable them to recognize and respond to distinct pathogens. Among these receptors, C-type lectins can be harnessed to deliver antigenic proteins to DCs or specific DC subsets. Accordingly, antigens can be introduced into mAbs that efficiently and specifically target to the C-type lectin receptor DEC in vivo13. DEC is highly expressed by DCs localized in lymphoid organs, specifically by a subset of DCs that also express CD8α14. DC targeting using DEC-specific mAbs (anti-DEC) induces efficient antigen processing and presentation on MHC class I and II products15,16,17. Notably, this strategy elicits both T helper 1 (TH1) CD4+ T-cell and CD8+ T-cell responses when mAb-antigen fusion proteins are administered in the presence of an adjuvant to maturate and/or differentiate DCs15,16,17,18.
Despite the advances made over the last decade in targeting antigens specifically to DCs, far less progress has been made with respect to the selective delivery of adjuvants. Typically, adjuvants are not targeted and consequently are recognized by a variety of cell types expressing PRRs (broad-acting adjuvants), which can lead to hyperactivation of innate immune mechanisms and adverse events19,20. In this study, we set out to examine whether DCs alone are able to immunize CD4+ and CD8+ T-cell responses after the targeting of both antigen and adjuvant. To do this, we adapted a protein engineering method, expressed protein ligation (EPL)21,22,23, to site-specifically attach a dsDNA adjuvant, pdA:dT, to an anti-DEC. Targeted delivery of pdA:dT to human monocyte–derived DCs (MoDCs) led to the production of type I IFNs in vitro. Similarly, we showed that in vivo delivery of pdA:dT to mouse DCs induced the secretion of type I IFNs. Finally, we demonstrate that mouse DCs activated with pdA:dT are able to immunize antigen-specific CD4+ and CD8+ T cells.
Results
Ligation of poly dA:dT to anti-DEC
The adjuvant of choice for our study was pdA:dT. This dsDNA is a potent activator of human MoDCs24 and was expected to be more stable than commonly used dsRNA adjuvants such as polyriboinosinic polyribocytidylic acid (poly I:C). To site-specifically conjugate pdA:dT to a full-length mAb, we developed a protein-DNA ligation strategy based on EPL. DNA for a modified intein compatible with protein secretion25 was cloned in frame into the C terminus of the heavy chain of anti–human DEC (anti-hDEC), anti−mouse DEC (anti-mDEC) and control immunoglobulin mAbs without receptor affinity (construct 1 in Fig. 1a). The resultant mAb-intein fusion proteins were produced by transient expression in 293T cells and were purified from the culture supernatants using protein G affinity chromatography16,17,26. SDS-PAGE (Supplementary Results, Supplementary Fig. 1a) and western blot analysis (Supplementary Fig. 1b) of the purified mAbs revealed that the preparation contained, in addition to the expected heavy chain–intein fusion proteins (∼75 kDa), a contaminant (∼50 kDa), which we attribute on the basis of its size to premature cleavage of the intein during the expression and/or purification protocol. Notably, attachment of the intein to the C terminus of the heavy chain did not disrupt antibody function because anti–DEC-intein bound as efficiently as unconjugated anti-DEC to the DEC receptor (Supplementary Fig. 1c).

(a) Schematic showing the chemoenzymatic generation of the antibody-DNA conjugates. Step i (thiolysis): the mAb-intein fusions (1) were treated with MESNA for 18 h to give anti-thioester derivatives (2). Step ii (ligation): the mAb-thioester derivatives (2) were mixed with 20 bp DNA oligo (dA:dT) containing a cysteine residue at the 5′ end of the dA oligo to give mAb–dA:dT conjugates (3). Step iii (elongation): purified mAb–dA:dT conjugates (3) were reacted with Klenow Pol-I fragment in the presence of dATP and dTTP to give anti–DEC-pdA:dT, 4. (b) Dose-dependent binding of anti–hDEC-pdA:dT (4 in a), to CHO cells expressing human DEC (CHO/hDEC; black) and control CHO/NEO (gray) cells as monitored by flow cytometry after staining with Phycoerythrin (PE)-conjugated anti–human IgG. Effect of hControl Ig-pdA:dT mAbs on the same cell lines is shown for comparison. (c) As in b, but binding was performed with mouse anti–mDEC-pdA:dT and CHO cells expressing mouse DEC (CHO/mDEC; black), after staining with PE-conjugated anti–mouse IgG.
The mAb-intein fusion proteins were next treated with sodium 2-mercaptoethanesulfonate (MESNA) to cleave off the intein and generate an active thioester at the C terminus of the heavy chain of each mAb (step i, resulting in protein derivative 2 in Fig. 1a). Approximately 24 h of treatment was necessary for efficient cleavage of the intein yielding a heavy chain of ∼50 kDa (Supplementary Fig. 1d,e). Reverse-phase liquid chromatography (RP-HPLC) and MS on the MESNA-treated deglycosylated and reduced anti–hDEC-thioester showed two peaks that corresponded to the expected light- and heavy-chain molecular weights (Supplementary Fig. 2a–c). Analysis by size-exclusion chromatography with in-line multi-angle light scattering analysis (SEC-MALS) showed that the mAb-thioester derivatives retained the expected tetrameric structure and did not aggregate in solution even after 2 weeks in storage at 4 °C (Supplementary Fig. 2d,e).
To evaluate the ability of anti–DEC-thioester derivate to undergo EPL, we first ligated a short fluorescent peptide. This approach allowed us to determine the amount of mAb-peptide fusion protein by measuring in-gel fluorescence. We found that by 24 h, more than 90% of the mAb-thioester was ligated to the short fluorescent peptide (Supplementary Fig. 3a). HPLC and MS analysis confirmed the generation of desired ligation product (molecular weight (MW) = 50,529.4 Da, calculated MW (MWcalcd) = 50,526.3 Da; Supplementary Fig. 3b). SEC-MALS analysis of the mAb-peptide fusion protein indicated that no aggregation had occurred during the thiolysis and ligation process (Supplementary Fig. 3c). Altogether, these model studies confirmed the capacity of anti–DEC-intein to undergo EPL without disrupting the antibody structure or adversely affecting protein behavior.
We next conjugated pdA:dT to anti–DEC-intein. It has been shown that at least 40 base pairs of dA:dT are necessary for immune activation, with the degree of activation steadily increasing as a function of DNA length beyond that threshold11,12. As initial efforts to directly ligate longer DNA molecules to the mAb-thioesters were unsuccessful, we adopted a two-step protocol. In the first step, we chemically ligated a short dsDNA oligonucleotide with an N-terminal cysteine (Cys-dsDNA; Supplementary Fig. 4)27,28 to the mAb-thioesters (step ii, resulting in ligation product 3 in Fig. 1a). To confirm ligation, we ran the mAb–dA:dT conjugates on a 4–20% Tris-borate-EDTA (TBE) native gel stained with ethidium bromide (Supplementary Fig. 5a). A band was visualized around 1,000 base pairs, which disappeared upon proteinase K treatment, confirming it was a mAb-DNA conjugate. The efficiency of dsDNA ligation was indirectly estimated using an in-gel fluorescence assay and ranged from 67% to 78% (Supplementary Fig. 5b). SEC-MALS analysis showed that mAb–dA:dT conjugates were of the appropriate size (MW = 185 kDa, MWcalcd = 172 kDa) and did not form aggregates after 24-h incubation at 22 °C (Supplementary Fig. 5c). In the final step, the dsDNA on the mAb–dA:dT was elongated using the Klenow polymerase I (step iii, resulting in construct 4 in Fig. 1a), which lacks exonuclease activity and adds extra bases through a slippage mechanism29. The reaction needed to proceed for ∼90 min to elongate the dsDNA to approximately 250 bp, which is the length of commercially available pdA:dT (Supplementary Fig. 5d)11,12. Notably, the thiolysis, ligation and extension procedures did not disrupt antibody function as anti–DEC-pdA:dT bound its corresponding receptor on stably transfected CHO cells similarly to unconjugated anti-DEC (Fig. 1b,c and Supplementary Fig. 1c).
Anti–hDEC-pdA:dT elicits type I IFN secretion from MoDCs
Cytoplasmic pdA:dT is a potent activator of human MoDCs for the production of type I IFN30. Previous reports indicate the need for dsDNA to access its intracellular targets, Pol-III and RIG-I, which can be accomplished by the use of transfection agents. Indeed, when we cultured MoDCs with different concentrations of soluble pdA:dT, IFN-α secretion was detected only when cells were also treated with the transfection agent lipofectamine (Fig. 2a).

(a) ELISA analysis of the production of IFN-α by MoDCs treated overnight with various doses of pdA:dT (with or without 1 μg ml−1 lipofectamine), anti–hDEC-pdA:dT and hControl Ig-pdA:dT. Error bars represent the mean ± s.d. of three independent experiments, each performed in triplicate. (b) Time course of the production of IFN-β by MoDCs treated with anti–hDEC-pdA:dT (5 μg ml−1) as determined by ELISA. Histograms represent mean ± s.d. (n = 3). (c) MoDCs were pretreated for 2 h with 10 μg of various blocking mAbs before adding 1 μg ml−1 or 5 μg ml−1 of anti–hDEC-pdA:dT. IFN-α production was monitored by ELISA and is shown as the mean ± s.d. of three experiments performed in triplicate per condition.
We next tested the ability of anti–hDEC-pdA:dT to trigger type I IFN production by human MoDCs. Accordingly, MoDCs were treated with different doses of either anti–hDEC-pdA:dT or human control (hControl) Ig-pdA:dT mAbs that have no receptor affinity. In contrast to the stimulation with soluble pdA:dT, anti–hDEC-pdA:dT induced the production of IFN-α without the use of any transfection reagent, suggesting that pdA:dT reached its intracellular target after DEC targeting (Fig. 2a). The IFN response was dependent on dose (Fig. 2a) and the time of exposure (Fig. 2b) and, consistent with a DEC receptor-mediated uptake mechanism, was not observed after treatment with hControl Ig-pdA:dT mAbs (Fig. 2a). Further, the response to anti–hDEC-pdA:dT was selectively inhibited by pretreatment of MoDCs with an excess of unconjugated anti-hDEC (Fig. 2c). These data indicate that delivery of pdA:dT to MoDCs using anti-hDEC induces the production of type I IFN without the need for transfection agents.
Activation of MoDCs by anti-hDEC-pdA:dT requires RIG-I
Intracellular pdA:dT is sensed by a pathway that involves RNA polymerase III and RIG-I11,12. To further explore the mechanism of immune activation after delivery of pdA:dT using anti-hDEC, we transfected human MoDCs with siRNA targeting MDA5 or RIG-I to knock down the expression of these nucleic acid sensors (Supplementary Fig. 6). As control stimuli, we used lipopolysaccharide (LPS) and poly I:C, which signal independently of these sensors, that is, through TLR4 and TLR3, respectively. Consistent with previously published data11,12, production of type I IFN (Fig. 3a,b) and TNF-α (Fig. 3c) by MoDCs transfected with poly dA:dT was greatly reduced in cells that had been treated with siRNA targeting RIG-I. Similarly, anti–hDEC-pdA:dT induced production of IFN-α and IFN-β, and TNF-α was also reduced in MoDCs treated with RIG-I–specific siRNA, whereas the MDA5 knockdown had no effect (Fig. 3a–c). Treatment of MoDCs with anti–hDEC-pdA:dT led to higher secretion of IFN-α and IFN-β than LPS and poly I:C (Fig. 3a,b) but less production of the proinflammatory cytokine TNF-α (Fig. 3c). Together, these results are consistent with a mechanism in which anti-hDEC deliver pdA:dT to the cytoplasm of human MoDCs, leading to the activation of RIG-I for the production of type I IFN.

(a–c) MoDCs transfected by electroporation with control, MDA5 or RIG-I specific siRNA were treated with 0.5 μg ml−1 of LPS, 5 μg ml−1 anti–hDEC-pdA:dT or 25 μg ml−1 poly I:C. Soluble pdA:dT (5 μg ml−1) was introduced into the cytoplasm using 1 μg ml−1 lipofectamine. Production of IFN-α (a), IFN-β (b) and TNF-α (c) was assessed by ELISA. Data is the mean ± s.d. of two experiments with triplicates per condition.
Anti–hDEC-pdA:dT triggers type I IFN production in vivo
Encouraged by the results in human MoDCs, we next tested our approach in vivo. It is known that mouse DCs transfected in vitro with pdA:dT release type I IFN11. To test whether pdA:dT delivered using anti-mDEC induced type I IFN production in vivo without the use of transfection agents, we treated B6 mice with different doses of anti–mDEC-pdA:dT (0.3–10 μg). IFN-α was detected in serum as soon as 3 h after inoculation of anti–mDEC-pdA:dT and was dose dependent (Fig. 4a). Anti–mDEC-pdA:dT produced similar amounts of IFN-α and IFN-β than soluble poly I:C (Fig. 4b,c), and this response was receptor mediated as it was greatly attenuated in DEC knockout mice treated with anti–mDEC-pdA:dT or in wild-type mice inoculated with a mouse control (mControl) Ig-pdA:dT mAb with no receptor affinity (Fig. 4b).

(a) Various doses of anti–mDEC-pdA:dT were inoculated intraperitoneally in B6 mice. Serum was collected at 3 h and 6 h and was then analyzed for IFN-α production using ELISA. Mean ± s.d. of two experiments with a total of three mice is shown. (b) B6, DEC knockout (KO) or Myd88/TRIF KO mice were inoculated intraperitoneally with the indicated immune stimulants (10 μg), and serum was analyzed for IFN-α production by ELISA. Shown is the mean ± s.d. of 2–5 experiments with 6–15 animals total. (c) B6 mice were inoculated intraperitoneally with indicated immune stimulants (10 μg), and serum was analyzed 1 h later for TNF-α production or 6 h later for IFN-α, IFN-β and IL-6 production by ELISA. Shown is the mean ± s.d. of two experiments with six animals total per group. In all cases, statistical differences were determined between all groups. ***P ≤ 0.001; NS, nonsignificant.
The sensor (or sensors) that detects pdA:dT and triggers type I IFN production in the mouse has remained largely unknown, but it seems that it is TLR independent7. To verify that the mechanism of action of anti–mDEC-pdA:dT was independent of any TLR pathway, we used Myd88- and TRIF-knockout mice (Myd88/TRIF), which lack two adaptor proteins necessary for this pathway3. As shown in Figure 4b, production of IFN-α triggered by inoculation with anti–mDEC-pdA:dT was comparable between Myd88/TRIF knockout and wild-type mice, suggesting that pdA:dT activates an innate response via a mechanism independent of TLRs. By contrast, the response to poly I:C was TLR dependent as it required the Myd88/TRIF pathway (Fig. 4b).
We further evaluated the advantage of targeting pdA:dT using anti-mDEC. As expected, mice inoculated with naked pdA:dT, administered in the absence of any transfection agent, failed to produce any serum cytokines (Fig. 4c). Also, production of type I IFN was significantly (P ≤ 0.001) more efficient when pdA:dT was delivered using anti-mDEC than when it was administered together with the transfection agent LyoVec (Fig. 4c). Remarkably, delivery of pdA:dT using anti-mDEC induced significantly smaller (P ≤ 0.001) amounts of proinflammatory cytokines, that is, TNF-α and IL-6 (Fig. 4c), and less hypothermia (Supplementary Fig. 7) than the same dose of untargeted poly I:C. Together these results suggest that anti-mDEC efficiently delivered pdA:dT to its targets for efficient production of type I IFN, causing less toxicity than the untargeted adjuvant poly I:C, as measured by production of proinflammatory cytokines and hypothermia.
DCs mediate type I IFN production by anti–mDEC-pdA:dT
As DEC is expressed at small amounts by other cells, including stromal cells, epithelial cells, B cells and granulocytes31, we evaluated the requirement of DCs for the in vivo production of type I IFN triggered by anti–mDEC-pdA:dT. Radio-resistant cells, such as endothelial, epithelial and/or stromal cells, but not bone-marrow derived hematopoietic cells (for example, DCs) are required for the bulk secretion of type I IFN in response to poly I:C18. To exclude a role for these nonhematopoietic cells in the production of IFN-α in response to anti–mDEC-pdA:dT, we generated bone marrow chimeras in which DEC was expressed only in radio-sensitive hematopoietic cells, including DCs (B6 wild-type bone marrow was transplanted into DEC knockout (KO) lethally irradiated mice; B6 → KO), or in radio-resistant cells (KO → B6). Twelve to fourteen weeks after reconstitution, chimeric mice were injected with anti–mDEC-pdA:dT labeled with Alexa 647, and the innate immune response was determined in the serum (Fig. 5a). Alexa 647–labeled anti–mDEC-pdA:dT was uptaken only by DCs from chimeric mice that received wild-type bone marrow (B6 → KO), corroborating proper chimerism (left panel in Fig. 5a). These chimeric mice (B6 → KO) also produced substantially more IFN-α (Fig. 5a) and IFN-β (Supplementary Fig. 8) compared to mice of the opposite chimerism (KO → B6), indicating that the majority of type I IFN was secreted by radio-sensitive hematopoietic cells expressing DEC, such as DCs, and not by radio-resistant stromal cells. To further investigate the requirement of DCs for the secretion of IFN-α triggered by anti–mDEC-pdA:dT inoculation, we performed experiments using CD11c-DTR mice. This transgenic strain allows for the depletion of DCs, but not B cells, T cells and granulocytes, after injection of diphtheria toxin32. As expected, diphtheria toxin–treated CD11c-DTR mice produced less IFN-α than mice not treated with diphtheria toxin, indicating that other leukocytes that expressed small amounts of DEC are dispensable for type I IFN production (Fig. 5b). Thus, the in vivo production of type I IFN triggered by anti–mDEC-pdA:dT is mediated by DCs and not radio-resistant cells or radio-sensitive leukocytes that express small amounts of DEC.

(a) B6 (wild type (WT)) or DEC knockout (KO) mice were lethally irradiated and injected with bone marrow cells from DEC KO mice (KO → B6) or B6 mice (B6 → KO), respectively. Chimeras and WT or DEC KO control mice were inoculated with 10 μg of Alexa 647–labeled anti–mDEC-pdA:dT. Spleens were harvested, and mAb uptake was evaluated by FACS in CD8α+ DCs (left). Bar graph (right) shows serum IFN-α as the mean ± s.d. of four mice in two experiments. (b) CD11c-DTR mice were inoculated with diphtheria toxin (+ DT) or PBS (– DT) 24 h before the inoculation of 10 μg of anti–mDEC-pdA:dT. ELISA was performed to determine serum IFN-α 6 h later. Shown is the mean ± s.d. of three animals per group. (c) B6 or DEC KO mice were inoculated intraperitoneally with 10 μg Alexa 647–labeled anti–mDEC-pdA:dT. Uptake of labeled mAbs by splenic DC subsets was evaluated by FACS. Results from one experiment of two is shown. (d) Spleens of mice inoculated with 10 μg of mAb–pdA:dT were harvested, and DC populations were FACS-sorted and cultured. IFN-α production was assessed by ELISA in the culture supernatant. Mean ± s.d. of two experiments is shown. (e) Surface costimulatory markers (CD86, MHCII and CD40) were evaluated 12 h after inoculation of 10 μg of anti–mDEC-pdA:dT or of PBS by FACS gating on the indicated DC populations. Fluorochrome-labeled isotype controls are shown in gray.
DCs are a heterogeneous population of leukocytes composed of several subsets with different functions and expression of surface receptors33. In lymphoid organs, the CD8α+ DC subset is characterized by high DEC expression and specializes in cross-presentation of antigen. To assess which subset of DCs captured anti–mDEC-pdA:dT in vivo, mAbs labeled with Alexa 647 were inoculated in wild-type or DEC knockout mice. As expected, anti–mDEC-pdA:dT labeled all splenic CD8α+ DCs in wild-type mice, but no labeling was observed in DEC knockout mice (Fig. 5c). To further examine whether CD8α+ DCs were the source of serum type I IFN production, we purified using fluorescence-activated cell sorting (FACS) and cultured distinct splenic DC subsets of mice treated with anti–mDEC-pdA:dT ex vivo for 12–18 h. Only CD8α+ DCs, but not CD8α− or plasmacytoid DCs (PDCs), were able to produce high amounts of type I IFN (Fig. 5d). Remarkably, upregulation of the costimulatory markers CD40 and CD86 and of MHCII was detected in CD8α+ DCs, CD8α− DCs and PDCs, suggesting that all DC subsets responded to the cytokines produced by the CD8α+ DCs (Fig. 5e).
Taken together, these results demonstrate that DCs expressing DEC are sufficient and responsible for the bulk secretion of type I IFN detected in the serum triggered by anti–mDEC-pdA:dT.
Targeting adjuvant and antigen to DCs activates T cells
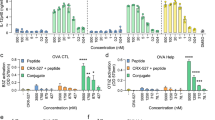
Next we asked whether mice could be immunized by directing both the antigen and the adjuvant primarily to DCs. To this end, 15 μg of anti–mDEC-pdA:dT was administered together with 5 μg of anti–mDEC-human immunodeficiency virus (HIV) Gag p24. The latter mAb efficiently immunizes mice against the HIV Gag p24 antigen when a broad-acting adjuvant, such as poly I:C, is also administered16,17,18. After prime-boost immunization, splenocytes were isolated, restimulated with a Gag p24–reactive peptide pool or a Gag p17–nonreactive peptide pool (negative control), and the frequency of CD4+ T cells producing IFN-γ was monitored by FACS. The frequency of IFN-γ–producing CD4+ T cells was higher when 7.5 μg of pdA:dT (equivalent to ∼15 μg of anti–mDEC-pdA:dT) were delivered using anti-mDEC compared to when 7.5 μg of soluble poly I:C were delivered (Fig. 6a). To exclude the possibility that mAbs conjugated with antigen and adjuvant would compete for their cognate receptor (DEC), we delivered the antigen to another receptor expressed on CD8α+ DCs called Langerin17. Anti–mouse Langerin (mLangerin)-Gag p24 administered in combination with anti–mDEC-pdA:dT was also able to prime antigen-specific CD4+ TH1 T-cell responses (Supplementary Fig. 9); however, the frequency of IFN-γ–producing cells was not statistically different than delivery of the antigen using anti-DEC (compare Fig. 6a with Supplementary Fig. 9).

(a) B6 mice were primed and boosted 4 weeks apart with 5 μg of HIV Gag p24 antigen fused to anti-mDEC. Fifteen micrograms of anti–mDEC-pdA:dT, 7.5–15 μg of poly I:C or 15 μg of mControl Ig-pdA:dT was used as the adjuvant. Seven days after the boost, splenocytes were restimulated in vitro with the reactive HIV Gag p24 peptide mix (p24) or a nonreactive HIV Gag p17 peptide pool (p17, negative control) in the presence of brefeldin A for 6 h. Intracellular staining was performed to detect IFN-γ in CD3+ CD4+ gated T cells. The percentage of IFN-γ+ CD4+ T cells is shown as the mean ± s.d. of three independent experiments with a total of six mice. (b) B6 mice were immunized with 5 μg anti–mLangerin-OVA in combination with 15 μg of either anti–mDEC-pdA:dT, 7.5–15 μg poly I:C or 15 μg of mControl Ig-pdA:dT mAbs. Two weeks after inoculation, IFN-γ production was evaluated by intracellular cytokine staining in the CD3+ CD8+ gated T cells in response to the reactive OVA peptide (OVAp). Shown is mean ± s.d. from two experiments with a total of 5 mice. In all cases, statistical differences were determined between mice inoculated with anti–mDEC-pdA:dT and the other groups. *P ≤ 0.05; NS, nonsignificant.
Finally, we determined whether anti–mDEC-pdA:dT was an effective adjuvant for CD8+ T-cell immunity, as there is no described epitope (or epitopes) for HIV Gag p24 in B6 mice (H-2b)34, we used ovalbumin (OVA) as our antigen, which contains the class I (Kb) restricted epitope OVA257–264 (peptide sequence SIINFEKL)35. Two weeks after immunization with a single dose of 5 μg anti–mLangerin-OVA26 in the presence of 15 μg anti–mDEC-pdA:dT, CD8+ T-cell priming was monitored by IFN-γ production after intracellular cytokine staining (Fig. 6b). The results indicated again that 15 μg of anti–mDEC-pdA:dT (equivalent to 7.5 μg of soluble pdA:dT) was more effective than 7.5 μg of soluble poly I:C at priming CD8+ T-cell immunity (Fig. 6b). These data demonstrate that dual delivery of antigen and adjuvant to DCs is an effective strategy for the priming of TH1 CD4+ and CD8+ T-cell responses in mice.
Discussion
Successful vaccines that elicit protective T-cell immunity contain not only protective antigen (or antigens) but also an adjuvant component that triggers innate immune activation and is necessary for immunogenicity2. Although antigens can be delivered specifically to DCs using a variety of uptake receptors, adjuvants often act broadly; that is, they activate a variety of cell types36. Herein, we evaluated whether DCs alone can integrate the innate and adaptive immune response when a dsDNA, pdA:dT, is delivered to them using mAbs against the surface receptor DEC. We found that anti–DEC-pdA:dT triggered the production of type I IFN by human MoDCs in vitro and in mice. Further, we showed that codelivery of adjuvant and antigen to DCs is able to bring about the induction of combined CD4 and CD8 T-cell immunity. Thus, targeted activation of DCs induces both innate and adaptive immunity in vivo.
To deliver an adjuvant to DCs, we developed a new chemoenzymatic route to a dsDNA-mAb conjugate using chemical ligation followed by polymerase-mediated DNA polymerization. The generation of DNA-antibody conjugates has been an active area of research since immuno-PCR was first described in 1992 (ref. 37). In this biosensing technology, the antibody is used to detect antigen, and the DNA moiety is used to increase sensitivity by PCR, amplifying the response by ∼1,000–10,000-fold over a standard ELISA38,39. Despite its obvious attractions, immuno-PCR is not widely used, in part because standard methods to create DNA-antibody conjugates lack precise chemical control over the site of DNA attachment, resulting in molecular heterogeneity40. The approach developed in the current study offers a route to DNA-antibody conjugates where there is defined stoichiometry and regiocontrol over the coupling site. The advantage of a site-specific attachment method is highlighted by a recent study in which random conjugation of the nucleic acid moiety CpG alters the binding of anti-DEC to its receptor41. Although expressed protein ligation has been used previously to generate protein-DNA conjugates42 and in the area of immunoconjugation28,43, this is to our knowledge the first time that a dsDNA, pdA:dT, has been ligated to a full-length mAb. One caveat to the use of EPL is premature hydrolysis of the intein from the protein of interest, in this case the antibody heavy chain. Indeed, intein self-hydrolysis was observed in the current study, which, depending on the preparation, was between 20–40% of the fusion. Although this reduced the yield and potency of the preparation, it does not seem to have altered its DC targeting properties—the anti–DEC-pdA:dT preparation worked as designed, despite containing a small amount of unconjugated antibody contaminant. Nonetheless, reducing the degree of intein hydrolysis would clearly be desirable for future studies. With respect to this, we have very recently developed a modified EPL protocol, using ultrafast split inteins, that eliminates premature intein cleavage, improving the yields of semisynthetic proteins, including antibodies44. We imagine that these EPL procedures can be adapted to the site-specific attachment of other nucleic acids to mAbs, and as a consequence they will have utility in immuno-PCR and related technologies. Moreover, we envision RNA (for example, siRNAs) and even small molecules being targeted to specific cells via conjugated mAbs created using this method.
The choice of dsDNA for conjugation to anti-DEC was based on several considerations. First, dsDNA is more stable than dsRNA, poly I:C being an example. Second, cytosolic dsDNA, for example, pdA:dT, is a potent activator of human myeloid DCs24,30, which, upon treatment, acquire the capacity to induce TH1 CD4+ and CD8+ cytotoxic T-cell responses24. Of note, it is not feasible to use soluble pdA:dT (pdA:dT naked) as a vaccine adjuvant in vivo, and transfection agents need to be used to improve access of dsDNA intracellular targets11,12. However, as we have shown, pdA:dT in complex with the transfection agent LyoVec is inefficient for production of type I IFN in vivo, at least under the dosing regime used in our study (conceivably higher doses of pdA:dT–LyoVec might elicit a stronger response). By contrast, the same dose of anti–DEC-pdA:dT can efficiently activate the production of type I IFN by DCs without the use of any transfection agent. A possible mechanism by which anti–DEC-pdA:dT activates in DCs is as follows (Supplementary Fig. 10). After entering the cells via DEC receptor–mediated endocytosis, pdA:dT is able to escape endosomes and reach its cytosolic targets. Notably, DCs have a unique ability to cross-present antigens, that is, to present extracellular antigens in MHC class I complexes45. The cellular mechanism of cross-presentation is still under debate, but it is possible that this involves the exit of antigen (or antigens) from the endosomal component to the cytoplasm46. Notably, cross-presentation is primarily mediated by the mouse CD8α+ DC subsets and their human equivalent, BDCA3+ DCs, which express large amounts of the surface receptor DEC47. We note that antigens delivered with anti-DEC are efficiently cross-presented in MHC I complexes15,17,48. In support of this model, our data with human MoDCs indicate that activation of the innate response is dependent on the RIG-I pathway, arguing that pdA:dT is delivered to the cytosol of DCs after DEC-mediated endocytosis. In mice, the mechanism of type I IFN production by pdA:dT remains unknown, although it has been shown to be RIG-I independent. Indeed, our results indicate that the production of type I IFN in mice was independent of Myd88/TRIF expression, excluding a role for TLR ligands in the activation mediated by anti–DEC-pdA:dT. Other DNA sensor (or sensors), such as ZBP1/DAI and LRRFIP1, may be responsible for pdA:dT recognition in our system11. Additional studies are necessary to elucidate the specific mechanism by which delivery of pdA:dT using anti-DEC activates the secretion of type I IFN; however, the reagents created in this study will be useful for dissecting this process.
PRRs are expressed on both immune and nonimmune cells, the latter including endothelial, epithelial and stromal cells3. Consequently, untargeted adjuvants usually activate a variety of cell types. For example, when mice are injected with poly I:C, large serum amounts of type I IFN are produced from nonimmune cells, such as stromal cells, which are essential for the immunization to take place18. In our experiments, delivery of pdA:dT to DCs was found to induce similar amounts of serum type I IFN as poly I:C. We demonstrated that radio-resistant cells, such as epithelial, endothelial and/or stromal cells, that express small amounts of DEC31 are not required for the bulk of type I IFN production elicited by anti–DEC-pdA:dT. Rather, in our targeting system, the initial release of type I IFN is mediated by DCs as it was abolished by administration of a control immunoglobulin mAb without receptor affinity in mice lacking expression of DEC and in those depleted of DCs. Notably, our adjuvant delivery system assures that DCs have direct recognition of the adjuvant, which is essential for their functional activation18,49, and demonstrates that DCs alone are capable of inducing adaptive immunity.
Various protein-based vaccine strategies, including approaches based on DCs targeting, are currently being evaluated in several preclinical settings13. A potential drawback with these existing strategies is the need to coadminister high doses of an untargeted broad-acting adjuvant. Such a formulation can lead to increased amounts of serum cytokines, which is often associated with toxicity19,20. Receptor-targeting methods may be a tool to decrease the toxicity and/or adverse events of adjuvants in vaccines, as shown recently by a nanoparticle encapsulation strategy for the codelivery of poly I:C, resiquimod and OVA to DCs50. Consistent with this, we found that anti–DEC-pdA:dT induced significantly (P values are in Fig. 4) less TNF-α and IL-6 than untargeted poly I:C. Furthermore, untargeted poly I:C induced a significantly higher degree of mice hypothermia than DEC-targeted pdA:dT. The simplest interpretation of these results is that targeting an adjuvant to DCs leads to less systemic toxicity. However, we cannot rule out that the observations might also stem from differences in dose response and/or mechanism of action between poly I:C and anti–mDEC205-pdA:dT. Additional studies will be required to tease this apart.
In summary, the current study is to our knowledge the first to demonstrate that production of type I IFN and induction of adaptive immune responses can be triggered by delivery of a dsDNA adjuvant only to DCs and excludes the role of other immune and nonimmune cells expressing lower amounts of DEC. As such, our semisynthetic strategy, which affords a distinct molecular entity, will potentially allow targeting of different adjuvants to distinct subsets of DCs for the induction of specific immune responses, that is, the TH1, TH2 and cytotoxic T-cell responses.
Methods
Reagents.
The following fluorescent conjugated anti-mouse mAbs were purchased from eBioscience (San Diego, CA) or BD Pharmingen (San Diego, CA): APC anti–IFN-γ (XMG1.2) and anti-CD11c (HL3), Alexa-488 anti–IL-2 (JES6-5H4), PE-Cy7 anti–TNF-α (MP6-XT22), eFluor 450 and Alexa-700 anti-CD3e (500A2), PerCP-Cy5.5 and Alexa-700 anti-CD4 (GK1.5), PerCP-Cy5.5 anti-CD8 (53-6.7), PE anti-CD40 (3/23), anti-CD86 (GL1), anti–MHC-II (IEk, 14-4-4S) and anti-CD11c (HL3). PE anti–PDCA-1 (JF05-1C2.4.1) was from Miltenyi Biotec (Auburn, CA). Live/Dead Fixable Aqua vitality dye was from Invitrogen, and DAPI was from Sigma (St. Louis, MO). Diphtheria toxin (DT), chloroquine and LPS were from Sigma-Aldrich (St. Louis, MO); human interleukin 4 (IL-4) was from R&D systems (Minneapolis, MN); and granulocyte-macrophage colony stimulating factor (GM-CSF) was from Berlex (Berkeley, CA). Poly I:C, pdA:dT Naked and pdA:dT LyoVec were obtained from InVivogen (San Diego, CA). Klenow DNA Pol-I fragment was purchased from New England BioLabs (Ipswich, MA). Amine-DNA oligomers were purchased from Fisher Scientific (Pittsburg, PA). Overlapping (staggered by four amino acids) 15-mer peptides covering the entire HIV Gag p17 or HIV Gag p24 or the OVA peptide (aa257-264, SIINFEKL) were synthesized by H. Zebroski in the Proteomics Resource Center at The Rockefeller University.
General equipment.
Size-exclusion chromatography (SEC) was carried out on ÄKTA Purifier system from GE Healthcare (Piscataway, NJ). Analytical SEC-MALS was carried out on a Superdex 200 10/300 column (GE Healthcare). RP-HPLC experiments were performed on Hewlett-Packard 1100 and 1200 series instruments using C18 or C4 Vydac columns (5 μm, 4.6 × 150 mm) at a flow rate of 1 ml/min. Modified oligonucleotides were analyzed using 10 mM TEAA (triethylammonium acetate) in water (solvent A) and 80% acetonitrile in water (solvent B). Anti-hDEC conjugates were analyzed using 0.025% TFA, 0.25% formic acid (FA) in water (solvent C) and 0.025% TFA, 0.25% FA, 90% acetonitrile in water (solvent D). ESI-MS analysis of anti-hDEC was performed on a MicrOTOF-Q II mass spectrometer from Bruker Daltonics (Billerica, MA). Multiple Angle Light Scattering (MALS) and Refractive Index (RI) measurements were performed using a Dawn Heleos II and Optilab T-rEX from Wyatt Technology Corporation (Santa Barbara, CA), respectively.
Construction of anti–receptor-intein.
Mycobacterium xenopi gyrase A intein (GyrA) was cloned in frame after the C terminus of anti-DEC heavy chain, analogous to that previously reported for fusion of immune antigens such as HIV Gag-p24 (refs. 16,17). Quikchange Site-Directed Mutagenesis Kit (Stratagene, Santa Clara, CA) was used to obtain the GyrA intein with Cys79 and Cys114 converted to serine. These mutations have been shown to improve the activity of the intein in the context of secreted protein fusions25. Using XhoI and NotI restriction sites incorporated into oligonucleotides primers, a DNA fragment containing the Mxe GyrA (C79S C114S) was cloned into pK::hDEC, a mammalian expression vector encoding both the heavy and light chain of human anti-DEC, to give the plasmid pK::hDEC-intein. The identity of the cloned gene was confirmed by DNA sequencing. HEK 293T cells were then transiently transfected with the pK::hDEC-intein vector using calcium phosphate in serum-free DMEM medium supplemented with Nutridoma SP (Roche, Indianapolis, IN). After 3 d, the medium was collected and filtered with Steritop vacuum filter cups (Millipore, Billerica, MA). The filtrate was loaded onto a column containing 1 ml of protein G sepharose beads (GE Healthcare, Piscataway, NJ). The beads were washed with 200 ml of Protein G binding buffer (Pierce, Rockford, IL), and the anti–DEC-Mxe GyrA (C79S C114S) intein mAb was eluted off the beads using Protein G elution buffer (Pierce, Rockford, IL). The eluent was dialyzed against phosphate buffered solution (PBS) and concentrated using an Amicon concentrator (Millipore, Billerica, MA). The typical yield for the preparation ranged from 1.25 mg/l to 2.5 mg/l. No effort was made to optimize these expression conditions. Similar cloning, expression and purification protocols were used to generate three additional mAb-intein fusions, namely; anti–mDEC-GyrA (C79S C114S) intein and two antibodies without receptor affinity, human control immunoglobulin- (hControl) intein and mouse control Ig- (mControl) intein.
Production of Cys-DNA.
A DNA oligonucleotide (sequence; AAAAAAAAAAAAAAAAAAAA) with a 5′ amine group was purchased from Fisher Scientific (Pittsburgh, PA). Fmoc-Cys(StBu)-OH (Sigma, St. Louis, MO) was activated with DIC and N-hydroxysuccinimide to generate Fmoc-Cys(StBu)-OSU. The amino-DNA was then reacted with 50-fold excess Fmoc-Cys(StBu)-OSU in a 1:1 solution of 50 mM borate buffer pH 8.5 and dimethylformamide (DMF). The protected Cys-DNA was purified by RP-HPLC using a C18 Vydac column where buffer A was 10 mM triethylammonium acetate (TEAA) in water and buffer B was 80% CH3CN in water. A gradient of 0–50% B over 30 min was used for purification. The purified protected Cys-DNA was incubated overnight in 30% ammonium hydroxide to remove the Fmoc protecting group. Finally, the sample was placed in 1 M dithiothreitol (DTT) to remove the S-t-butyl protecting group. The deprotected Cys-DNA was purified using the same HPLC gradient as before. The identity of the product was confirmed by MALDI-TOF MS; 6,483.4 Da (calculated); 6,495.7 Da (observed).
Expressed protein ligation.
An oligonucleotide of sequence TTTTTTTTTTTTTTTTTTTT was purchased from Fisher Scientific (Pittsburgh, PA). This was combined in a 1:1 ratio with Cys-DNA in water, and the strands were annealed by heating to 95 °C for 5 min followed by gradual cooling to room temperature for 1 h. Antibody-intein fusion proteins were activated by thiolysis through the addition of 100 mM sodium 2-mercaptoethanesulfonate (MESNA) (Sigma, St. Louis, MO) for 18 h in PBS (phosphate-buffered saline). The resulting antibody–anti-thioester derivatives (50 μM) were reacted with the double stranded (ds)-Cys-DNA (500 μM) in ligation buffer (1× PBS, 400 mM NaCl, 6 mM TCEP, 1 mM EDTA, 100 mM MESNA, pH 7.5). The reactions were typically allowed to proceed at room temperature overnight. Reaction progress was monitored by gel electrophoresis using 4–20% TBE native gels (Bio-Rad, Hercules, CA), using ethidium bromide (Invitrogen, Carlsbad, CA) staining to check for ligation. Crude ligation mixtures were then dialyzed five times against PBS for at least 2 h at 4 °C using Slide-A-Lyzer Cassettes (20,000 MWCO; Pierce, Rockford, IL). The conjugates were then concentrated using 100,000-MWCO microcon concentrators (Millipore, Billerica, MA). Trace amounts of Cys-DNA were usually still seen at this point, as visualized on the 4–20% TBE native gels stained with EtBr. If necessary, the samples were diluted to 1 ml and reconcentrated (5 mg/ml) until no Cys-DNA was visible on the EtBr gel (usually two times).
Elongation.
Purified mAb-DNA conjugates (3–4 μM) were reacted with 5 μM Klenow fragment in the presence of 0.3 mM dATP and dTTP for 90 min in NEB buffer 2 (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT). Elongation progress was monitored by treating an aliquot of the reaction mixture with 0.1 μg/ml proteinase K (Qiagen, Valencia, CA) for 1 h followed by analysis on a 2% agarose gel stained with EtBr (Supplementary Fig. 5d). Samples were then purified and concentrated using 100,000-MWCO microcon concentrators (Millipore, Billerica, MA). This procedure was used to produce site-specific pdA:dT conjugates to both the human and mouse anti-DEC as well as to the human control immunoglobulin and the mouse control immunoglobulin mAbs.
RP-HPLC analysis of anti–hDEC-thioester and derivatives.
Thirty to fifty micrograms of anti-hDEC were typically used for RP-HPLC/MS analysis. Before RP-HPLC analysis, mAbs were fully deglycosylated and fully reduced51,52. Deglycosylation was performed using 2 U of PNGase F (New England Biolabs) per μg of mAbs and incubated at 37 °C overnight. Next, mAbs were denatured by exchanging buffer to 6 M Gn·HCl, 100 mM phosphate, 150 mM NaCl, 1 mM EDTA at pH 7.2 and were fully reduced by treatment with 10 mM DTT at 37 °C for 1 h. The fully denatured and reduced anti-hDEC mAb was analyzed by RP-HPLC on a C4 Vydac column (5 μm, 4.6 × 150 mm) using a 5–75% linear gradient of solvent D in solvent C over 30 min at 1 ml/min flow rate and 55 °C, preceded by a 5-min isocratic phase at 5% D. Solvent C is 0.02% TFA and 0.25% FA (formic acid) in water, and solvent D is 90% isopropanol in water with 0.02% TFA and 0.25% FA. Peaks were collected and analyzed by ESI-MS.
SEC-MALS of anti–hDEC-thioester and derivatives.
The integrity of modified anti-hDEC was confirmed by injecting 25–50 μg of peptide- or dA:dT-ligated and PBS-dialyzed mAb in running buffer (100 mM phosphate, 150 mM NaCl, 1 mM EDTA, 1 mM TCEP, pH 7.2) into an S200 size-exclusion column using the ÄKTA Purifier system. Elution from the column was monitored by UV-visible absorbance at 280 nm, 495 nm and 254 nm and by online Dawn Heleos II MALS and Optilab T-rEX RI detectors. MALS, UV-Vis and RI data were analyzed using the ASTRA software. The measured polydispersity index value close to 1.00 for each anti-hDEC derivative tested indicated the tetrameric IgG remained monodisperse after thiolysis from the intein and ligation.
Estimation of anti–DEC-pdA:dT yields.
Approximately, 1.25–2.5 mg of anti–DEC-intein was purified per liter of transfection supernatant. SDS-PAGE and Coomassie blue staining was then performed to determine the extent of intein self-hydrolysis in the preparation. Typically, ∼20–40% of the sample was self-hydrolyzed during preparation. Hydrolyzed mAb is unable to react and therefore was not used in the calculation of moles of mAb for ligation. It was then assumed that MESNA treatment of the sample went to a 100% completion. For ligation, 100 μL of 50 μM mAb-thioester was used (5 nmol). Ligation efficiency was indirectly determined using the fluorescence assay shown in Supplementary Figure 5b. Typically, ∼60–70% of the mAb-thioester reacted (3–3.5 nmol; Supplementary Fig. 5b). It was then approximated that elongation of the short DNA attached to the antibody went to 100% (Supplementary Fig. 5d). During preparation, 30–50% of the antibody was lost (during concentration and purification). Thus, the final yield was typically around 1 nmol/l or 200 μg/l of initial culture.
Integrity and quality control of anti–receptor-ds DNA fusion.
mAbs were characterized by SDS/PAGE and western blotting using anti–human IgG or anti–mouse IgG conjugated with HRP (Jackson ImmunoResearch, West Grove, PA). mAb binding to their cognate receptor was verified on CHO cells stably transfected with their respective receptor. In brief, CHO cells stably transfected to express hDEC or mDEC receptors and control CHO NEO cells without receptor expression were incubated with either anti-human or mouse DEC-dsDNA or control Ig-dsDNA conjugates (2 μg, 0.2 μg and 0.02 μg) at 4 °C. Cells were washed with FACS buffer containing 2 mM EDTA (FACS/EDTA), after which either anti–human IgG or anti–mouse IgG (depending on antibody type) conjugated with PE was added. After incubation for 20 min at 4 °C, the cells were washed with FACS/EDTA buffer. Flow cytometry was then performed to determine the extent of anti–hDEC-pdA:dT and anti–mDEC-pdA:dT binding to their corresponding receptors.
Human cell isolation and culturing conditions.
Human PBMCs were isolated from whole blood of healthy volunteers by density gradient centrifugation using Ficoll-Hypaque (GE Healthcare, Piscataway, NJ). Human monocytes were isolated from PBMCs with anti-CD14 paramagnetic beads (Miltenyi Biotec, Auburn, CA) and were differentiated for 5 d into MoDCs in the presence of IL-4 (20 ng/ml) and GM-CSF (20 ng/ml). Primary cells were cultured in RPMI (GIBCO, Carlsbad, CA) medium supplemented with 2% pooled human serum (GemCell, Pennant Hills, NSW). For stimulation assays, 1 × 106 cells were plated in a 96-well-bottom plate and incubated with 0.5–5 μg/ml anti–hDEC-pdA:dT or a hControl Ig-pdA:dT mAb for 3–24 h. In some experiments, cells were pretreated with 10 μg/ml of purified mAbs against MHCII, CD11c or DEC205 or a control IgG. In all cases, supernatant was collected, and human INF-α, human IFN-β (PBL Interferon Source, Piscataway, NJ) or human TNF-α (R&D systems, Minneapolis, MN) were assessed with commercial ELISA kits according to the manufacturer's instructions.
Electroporation of human MoDCs with siRNA.
All siRNA sequences were purchased from Sigma-Aldrich (St. Louis, MO). The RIG-I siRNA sequence was 5′-AAGGCUGGUUCCGUGGCUUUUdTdT-3′. Three MDA5 siRNA sequences were used on the basis of a previous study53: 5′-GUUCAGGAGUUAUCGAACAdTdT-3′, 5′-GUAACAUUGUUAUCCGUUAdTdT-3′ and 5′-GGUGUAAGAGAGCUACUAAdTdT-3′. The control siRNA sequence (targets luciferase) was 5′-CUUACGCUGAGUACUUC GAdTdT-3′. Human MoDCs (4 × 106, at a concentration of 4 × 107/ml) were electroporated with 1 nmol of siRNA in opti-MEM without phenol red (Invitrogen, Carlsbad, CA). Electroporation was performed in a 4-mm cuvette and in a total volume of 200 μL of opti-MEM. Cells were pulsed (a unique square wave pulse of 500 V and 0.5 ms) using the ECM830 Electro Square Porator (BTX Harvard Apparatus). Immediately after electroporation, cells were transferred in complete medium (RPMI 2% human serum) supplemented with GM-CSF (20 ng/ml) and IL-4 (20 ng/ml). 24 h after electroporation, human MoDCs were collected and lysed with TRIzol (Invitrogen, Carlsbad, CA) followed by one round of chloroform extraction. Total RNA was precipitated with isopropanol and washed with 75% ethanol. Reverse transcription was then performed on isolated RNA using High Capacity RNA-to-cDNA Kit (Applied Biosystems, Carlsbad, CA). RT-PCR conditions and primer sequences for RIG-I and MDA-5 were previously described53.
Mice.
C57BL/6 (B6) mice were obtained from The Jackson Laboratory. DEC KO and Myd88/TRIF KO mice were kindly provided by M. Nussenzweig (The Rockefeller University, New York) and E.G. Pamer (Memorial Sloan-Kettering Cancer Center), respectively. CD11c-DTR mice were obtained from The Jackson Laboratory and bred in house. Mice were maintained under specific pathogen-free conditions and used at 7–8 weeks of age according to The Rockefeller University Animal Care and Use guidelines. To generate bone marrow chimeric mice, B6 or DEC KO mice were lethally irradiated twice 3 h apart with 550 rad and 600 rad, respectively. Three hours after the last irradiation, 3–5 × 106 bone marrow cells from either DEC KO (DEC KO into B6; KO → B6) or B6 (B6 into DEC KO; B6 → KO) were given intravenously. Mice were maintained in antibiotic food and water for at least 1 month after irradiation. Reconstitution was assessed at around 8 weeks by flow cytometry of blood cells (CD45+ leukocytes >98% donor origin), and mice were used 12–14 weeks after bone marrow transplant.
Mice inoculations and Immunizations.
To evaluate in vivo cytokine secretion, B6, DEC KO or MyD88/TRIF KO mice were injected intraperitoneally with different amounts of anti–mDEC205-pdA:dT (0.3–10 μg) or 10 μg of untargeted poly I:C, pdA:dT naked or pdA:dT LyoVec. Serum was collected 3–12 h after inoculation, and body temperature was measured using a microtherma 2T thermometer (ThermoWorks). Production of IFN-α (PBL Interferon Source, Piscataway, NJ), IFN-β (PBL Interferon Source, Piscataway, NJ), TNF-α (R&D systems, Minneapolis, MN) and IL-6 (R&D systems, Minneapolis, MN) was determined by ELISA following manufacturer's instructions. To evaluate capture of anti–mDEC-dsDNA fusion by DCs, anti–mDEC205-pdA:dT was labeled with Alexa 647 (Invitrogen Carlsbad, CA) per the manufacturer's instructions. B6 or DEC KO mice were injected intraperitoneally with 10 μg of Alexa 647–labeled mAbs, and uptake was evaluated 3 h after inoculation by multicolor flow cytometry after enzymatic digestion of the spleens (gating strategy described in ref. 17). In some experiments, spleens from mice inoculated for 12 h with anti–mDEC-pdA:dT were collected, digested enzymatically and monitored by multicolor flow cytometry for expression of several surface markers including MHCII (I-Ek), CD86 and CD40 in splenic DC subsets, including plasmacytoid DCs (CD11clow and PDCA-1+), CD11chigh CD8α+ DCs and CD11chigh CD8α− DCs (gating strategy described in ref. 17). For immunizations, B6 mice were inoculated in a prime-boost regimen that consisted in two intraperitoneal inoculations, separated by 1 month, of 5 μg anti-receptor mAbs fused to HIV Gag-p24 (anti-mDEC or anti–Langerin-Gag p24)17 in the presence of an adjuvant, which was 7.5–15 μg of soluble poly I:C (InVivogen) or 15 μg of anti–mDEC205-pdA:dT. In other experiments, mice were immunized intraperitoneally once with 5 μg anti–Langerin-OVA26 with either 7.5–15 μg of soluble poly I:C or 15 μg anti–mDEC-pdA:dT as a stimulus for DC maturation.
Cell sorting.
Spleens from B6 mice inoculated with 15 μg of anti–mDEC205-pdA:dT or mControl Ig-pdA:dT mAbs were cut in small pieces and incubated at 37 °C for 30 min in Hank's medium supplemented with 400 U/ml Collagenase D (Roche Diagnostics, Indianapolis, IN) and 50 μg/ml DNase I (Roche). EDTA (5 mM) was added for the last 5 min. Cells were washed, and red blood cells were lysed for 1 min using RBC lysis buffer (eBioscience, San Diego, CA). Cells were washed and stained with CD8α, CD11c and Live/Dead Fixable Aqua viability dye. Different cell populations were isolated by sorting on a FACSAria III system. Three populations, CD11clow DCs, CD8α+ DCs and CD8α- DCs (gating strategy described in ref. 18), were then plated at 300,000 cells per well in RPMI supplemented with 5% FCS (Gibco). Twelve to eighteen hours later, the supernatant was collected, and IFN-α secretion was evaluated by ELISA (PBL Interferon Source, Piscataway, NJ) following manufacturer's instructions.
Intracellular cytokine staining.
Bulk splenocytes were restimulated with the entire reactive HIV Gag-p24 or nonreactive negative HIV Gag-p17 peptide mix (peptides at 1 μg/ml) or with 2 μg/ml OVA peptide (SIINFEKL), along with 2 μg/ml of co-stimulatory anti-CD28 (clone 37.51) for 6 h at 37 °C. Brefeldin A (10 μg/ml; Sigma-Aldrich, St. Louis, MO) was added for the last 5 h to allow accumulation of intracellular cytokines. Cells were washed, incubated for 10 min at 4 °C with 2.4G2 mAbs to block Fcγ receptors, washed and stained with a cocktail of mAbs including Live/Dead Fixable Aqua viability dye, anti-CD3, anti-CD4 and anti-CD8 mAbs for 20 min at 4 °C. Cells were then fixed, permeabilized (Cytofix/Cytoperm Plus; BD Biosciences, Bedford, MA) and stained with anti–IFN-γ. A total of 1–3 × 105 live CD3+ cells were acquired on a Becton Dickinson LSR II flow cytometer, and data were analyzed with FlowJo Software (Tree Star, Inc., San Carlos, CA).
Statistical analysis.
Data reported in the figures were analyzed and charts were generated using Prism 5 (GraphPad Software). Statistical significance between two groups was determined by a Student's t-test. Statistical significance between more than two groups was determined by ANOVA followed by Bonferroni's post hoc analysis. In the figures, P values of ≤0.05 are labeled with a single asterisk (*), in contrast to P values of ≤0.01 (**) or ≤0.001 (***).
Additional information
Supplementary information is available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Correspondence and requests for materials should be addressed to T.W.M.
References
Ebensen, T. & Guzman, C.A. Immune modulators with defined molecular targets: cornerstone to optimize rational vaccine design. Hum. Vaccin. 4, 13–22 (2008).
Pulendran, B. & Ahmed, R. Translating innate immunity into immunological memory: implications for vaccine development. Cell 124, 849–863 (2006).
Kawai, T. & Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 (2011).
Yoneyama, M. & Fujita, T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 227, 54–65 (2009).
Iwasaki, A. & Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 327, 291–295 (2010).
Kawai, T. & Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21, 317–337 (2009).
Barber, G.N. Cytoplasmic DNA innate immune pathways. Immunol. Rev. 243, 99–108 (2011).
Stetson, D.B. & Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103 (2006).
Okabe, Y., Kawane, K., Akira, S., Taniguchi, T. & Nagata, S. Toll-like receptor–independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J. Exp. Med. 202, 1333–1339 (2005).
Ishii, K.J. et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7, 40–48 (2006).
Ablasser, A. et al. RIG-I–dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072 (2009).
Chiu, Y.H., Macmillan, J.B. & Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 (2009).
Trumpfheller, C. et al. Dendritic cell–targeted protein vaccines: a novel approach to induce T-cell immunity. J. Intern. Med. 271, 183–192 (2012).
Idoyaga, J., Suda, N., Suda, K., Park, C.G. & Steinman, R.M. Antibody to Langerin/CD207 localizes large numbers of CD8α+ dendritic cells to the marginal zone of mouse spleen. Proc. Natl. Acad. Sci. USA 106, 1524–1529 (2009).
Bonifaz, L.C. et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 199, 815–824 (2004).
Trumpfheller, C. et al. Intensified and protective CD4+ T cell immunity in mice with anti-dendritic cell HIV gag fusion antibody vaccine. J. Exp. Med. 203, 607–617 (2006).
Idoyaga, J. et al. Comparable T helper 1 (TH1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc. Natl. Acad. Sci. USA 108, 2384–2389 (2011).
Longhi, M.P. et al. Dendritic cells require a systemic type I interferon response to induce CD4+ TH1 immunity with poly IC as adjuvant. J. Exp. Med. 206, 1589–1602 (2009).
Lampkin, B.C., Levine, A.S., Levy, H., Krivit, W. & Hammond, D. Phase II trial of a complex polyriboinosinic-polyribocytidylic acid with poly-l-lysine and carboxymethyl cellulose in the treatment of children with acute leukemia and neuroblastoma: a report from the Children's Cancer Study Group. Cancer Res. 45, 5904–5909 (1985).
Batista-Duharte, A., Lindblad, E.B. & Oviedo-Orta, E. Progress in understanding adjuvant immunotoxicity mechanisms. Toxicol. Lett. 203, 97–105 (2011).
Muir, T.W., Sondhi, D. & Cole, P.A. Expressed protein ligation: a general method for protein engineering. Proc. Natl. Acad. Sci. USA 95, 6705–6710 (1998).
Muir, T.W. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 72, 249–289 (2003).
Muralidharan, V. & Muir, T.W. Protein ligation: an enabling technology for the biophysical analysis of proteins. Nat. Methods 3, 429–438 (2006).
Kis-Toth, K., Szanto, A., Thai, T.H. & Tsokos, G.C. Cytosolic DNA-activated human dendritic cells are potent activators of the adaptive immune response. J. Immunol. 187, 1222–1234 (2011).
Singla, N., Himanen, J.P., Muir, T.W. & Nikolov, D.B. Toward the semisynthesis of multidomain transmembrane receptors: modification of Eph tyrosine kinases. Protein Sci. 17, 1740–1747 (2008).
Idoyaga, J. et al. Langerin/CD207 receptor on dendritic cells mediates efficient antigen presentation on MHC I and II products in vivo. J. Immunol. 180, 3647–3650 (2008).
Lovrinovic, M. & Niemeyer, C.M. Rapid synthesis of DNA-cysteine conjugates for expressed protein ligation. Biochem. Biophys. Res. Commun. 335, 943–948 (2005).
Burbulis, I., Yamaguchi, K., Yu, R., Resnekov, O. & Brent, R. Quantifying small numbers of antibodies with a 'near-universal' protein-DNA chimera. Nat. Methods 4, 1011–1013 (2007).
Kotlyar, A.B., Borovok, N., Molotsky, T., Fadeev, L. & Gozin, M. In vitro synthesis of uniform poly(dG)-poly(dC) by Klenow exo- fragment of polymerase I. Nucleic Acids Res. 33, 525–535 (2005).
Katashiba, Y. et al. Interferon-α and interleukin-12 are induced, respectively, by double-stranded DNA and single-stranded RNA in human myeloid dendritic cells. Immunology 132, 165–173 (2011).
Witmer-Pack, M.D., Swiggard, W.J., Mirza, A., Inaba, K. & Steinman, R.M. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. II. Expression in situ in lymphoid and nonlymphoid tissues. Cell Immunol. 163, 157–162 (1995).
Bar-On, L. & Jung, S. Defining in vivo dendritic cell functions using CD11c-DTR transgenic mice. Methods Mol. Biol. 595, 429–442 (2010).
Belz, G.T. & Nutt, S.L. Transcriptional programming of the dendritic cell network. Nat. Rev. Immunol. 12, 101–113 (2012).
Mata, M., Travers, P.J., Liu, Q., Frankel, F.R. & Paterson, Y. The MHC class I–restricted immune response to HIV-gag in BALB/c mice selects a single epitope that does not have a predictable MHC-binding motif and binds to Kd through interactions between a glutamine at P3 and pocket D. J. Immunol. 161, 2985–2993 (1998).
Rötzschke, O. et al. Exact prediction of a natural T cell epitope. Eur. J. Immunol. 21, 2891–2894 (1991).
Nolte, M.A., LeibundGut-Landmann, S., Joffre, O. & Reis e Sousa, C. Dendritic cell quiescence during systemic inflammation driven by LPS stimulation of radioresistant cells in vivo. J. Exp. Med. 204, 1487–1501 (2007).
Sano, T., Smith, C.L. & Cantor, C.R. Immuno-PCR: very sensitive antigen detection by means of specific antibody-DNA conjugates. Science 258, 120–122 (1992).
Hendrickson, E.R., Truby, T.M., Joerger, R.D., Majarian, W.R. & Ebersole, R.C. High sensitivity multianalyte immunoassay using covalent DNA-labeled antibodies and polymerase chain reaction. Nucleic Acids Res. 23, 522–529 (1995).
McKie, A., Samuel, D., Cohen, B. & Saunders, N.A. Development of a quantitative immuno-PCR assay and its use to detect mumps-specific IgG in serum. J. Immunol. Methods 261, 167–175 (2002).
Niemeyer, C.M. Semisynthetic DNA-protein conjugates for biosensing and nanofabrication. Angew. Chem. Int. Ed. Engl. 49, 1200–1216 (2010).
Kreutz, M. et al. Antibody-antigen-adjuvant conjugates enable co-delivery of antigen and adjuvant to dendritic cells in cis but only have partial targeting specificity. PLoS ONE 7, e40208 (2012).
Niemeyer, C.M., Wacker, R. & Adler, M. Combination of DNA-directed immobilization and immuno-PCR: very sensitive antigen detection by means of self-assembled DNA-protein conjugates. Nucleic Acids Res. 31, e90 (2003).
Möhlmann, S., Bringmann, P., Greven, S. & Harrenga, A. Site-specific modification of ED-B–targeting antibody using intein-fusion technology. BMC Biotechnol. 11, 76 (2011).
Vila-Perelló, M. et al. Streamlined expressed protein ligation using split inteins. J. Am. Chem. Soc. 135, 286–292 (2013).
Flinsenberg, T.W., Compeer, E.B., Boelens, J.J. & Boes, M. Antigen cross-presentation: extending recent laboratory findings to therapeutic intervention. Clin. Exp. Immunol. 165, 8–18 (2011).
Lin, M.L., Zhan, Y., Villadangos, J.A. & Lew, A.M. The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol. Cell Biol. 86, 353–362 (2008).
Ueno, H. et al. Harnessing human dendritic cell subsets for medicine. Immunol. Rev. 234, 199–212 (2010).
Bozzacco, L. et al. DEC-205 receptor on dendritic cells mediates presentation of HIV gag protein to CD8+ T cells in a spectrum of human MHC I haplotypes. Proc. Natl. Acad. Sci. USA 104, 1289–1294 (2007).
Spörri, R. & Reis e Sousa, C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat. Immunol. 6, 163–170 (2005).
Tacken, P.J. et al. Targeted delivery of TLR ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 118, 6836–6844 (2011).
Wagner-Rousset, E. et al. The way forward, enhanced characterization of therapeutic antibody glycosylation: comparison of three level mass spectrometry-based strategies. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 872, 23–37 (2008).
Dillon, T.M. et al. Optimization of a reversed-phase high-performance liquid chromatography/mass spectrometry method for characterizing recombinant antibody heterogeneity and stability. J. Chromatogr. A 1120, 112–120 (2006).
Sasai, M. et al. NAK-associated protein 1 participates in both the TLR3 and the cytoplasmic pathways in type I IFN induction. J. Immunol. 177, 8676–8683 (2006).
Acknowledgements
The authors thank members of the Steinman and Muir laboratories for many valuable discussions. Funding was provided by National Institutes of Health Grants GM086868 (to T.W.M.), AI013013 (to R.M.S.), a Foundation for the National Institutes of Health Gates Foundation grant no. 334 (to R.M.S.) and a graduate fellowship to S. Barbuto by NIH MSTP grant GM07739.
Author information
Authors and Affiliations
Contributions
R.M.S. and T.W.M. conceived the original idea and contributed equally to this manuscript. T.W.M., S.B. and M.V.-P. contributed to the design and discussion of the chemical work; R.M.S., S.B. and J.I. contributed to the design and discussion of the biological work; S.B. and M.V.-P. synthesized and characterized mAb–pdA:dT conjugates; S.B. performed experiments with MoDCs with help from G.B.; S.B. and J.I. performed experiments in vivo with help and advice from M.P.L.; S.B., J.I. and M.V.-P. analyzed data and prepared figures; S.B., J.I., M.V.-P. and T.W.M. wrote the manuscript. All authors edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Results (PDF 2298 kb)
Rights and permissions
About this article
Cite this article
Barbuto, S., Idoyaga, J., Vila-Perelló, M. et al. Induction of innate and adaptive immunity by delivery of poly dA:dT to dendritic cells. Nat Chem Biol 9, 250–256 (2013). https://doi.org/10.1038/nchembio.1186
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nchembio.1186
This article is cited by
-
Template-directed covalent conjugation of DNA to native antibodies, transferrin and other metal-binding proteins
Nature Chemistry (2014)
-
Distribution of Antigen-Presenting Cells CD68 in Papillomavirus Infection in the Skin
Bulletin of Experimental Biology and Medicine (2014)
