
Abstract
The development of high-throughput sequencing technologies has advanced our understanding of cancer. However, characterizing somatic structural variants in tumor genomes is still challenging because current strategies depend on the initial alignment of reads to a reference genome. Here, we describe SMUFIN (somatic mutation finder), a single program that directly compares sequence reads from normal and tumor genomes to accurately identify and characterize a range of somatic sequence variation, from single-nucleotide variants (SNV) to large structural variants at base pair resolution. Performance tests on modeled tumor genomes showed average sensitivity of 92% and 74% for SNVs and structural variants, with specificities of 95% and 91%, respectively. Analyses of aggressive forms of solid and hematological tumors revealed that SMUFIN identifies breakpoints associated with chromothripsis and chromoplexy with high specificity. SMUFIN provides an integrated solution for the accurate, fast and comprehensive characterization of somatic sequence variation in cancer.
Similar content being viewed by others


Main
The recent development of high-throughput sequencing technologies has made possible the sequencing of genomes at an unprecedented speed, allowing the identification of the genetic basis of numerous diseases. These advances have been particularly important in the study of cancer, providing information on thousands of tumor genomes and a large catalog of genomic alteration associated with oncogenesis1. The characterization of somatic variation in tumor samples is, therefore, rapidly becoming a standard practice in biomedicine2. In a large fraction of biomedical studies that rely on high-throughput sequencing, the production of genome sequence data exceeds available computer resources and the capabilities of analytic protocols. This is particularly pertinent in the field of cancer genomics, where the increasing sequencing of tumor genomes calls for faster and more accurate analyses.
The identification of somatic variants associated with cancer typically requires sequencing tumor and normal genome samples from the same patient, followed by multiple sequence comparisons. Normal and pathological reads are aligned to a reference genome, and the alignment is used to identify sequence changes to isolate the somatic fraction of variants (i.e., those detected only in the tumor). In principle, this simple strategy can be used to detect single-nucleotide variants (SNVs) and structural variants. Existing methods for the detection of somatic SNVs show high sensitivity and specificity3,4, but identifying structural variants is still challenging and remains largely unsolved. The need for a reference sequence is particularly limiting. Reads carrying variations, such as those covering somatic changes in the tumor, are more difficult to align to the reference genome5, and corresponding variants might become undetectable. Moreover, reference-based methods also must discriminate germline changes from somatic variants. In addition to these limitations at detection level, this alignment step is also time consuming and requires a considerable amount of computing resources.
To define the complete catalog of somatic variation (SNVs and structural variants) for a given tumor still requires complex computational pipelines with combinations of different methods, each of them restricted to the detection of a particular type of variant or structural variants of particular sizes. This restricts the general usage of this methodology to centers and groups with considerable amounts of computing resources and expertise. For example, widely used programs, such as BreakDancer6 or Delly7, can only identify structural variants larger than 20 and 150 base pairs, respectively. Each of the methods needed for a complete structural characterization of somatic variation in tumor genomes further require complex scoring and filtering schemes to achieve acceptable levels of specificity, but such procedures drastically lower the sensitivity, leaving a substantial fraction of structural variants undetected. Even experimental procedures, such as those that use single-nucleotide polymorphism (SNP) arrays, generate only a partial description of the rearranged tumor, as they detect only the fraction of structural variation that generates sequence imbalance. The fact that the most recent and complete catalog for signatures of somatic mutations in cancer8 does not yet include structural variants is a clear consequence of all these limitations.
To fill these gaps, we have developed SMUFIN (for somatic mutation finder), a computational approach for the accurate and complete characterization of somatic variation in cancer. SMUFIN searches for SNVs and structural variants of all sizes by directly comparing normal and tumor sequencing reads without the need of their initial mapping onto a reference genome. Here, we evaluated its performance in the context of existing strategies and the application to cancer genomics, as well as its potential to define complex chromosomal rearrangements in aggressive forms of mantle cell lymphomas and medulloblastoma. The implementation of SMUFIN, including latest releases, documentation, example data sets and supplementary information is freely available at http://cg.bsc.es/smufin/. Source code files are also in Supplementary Source Code.
Results
The SMUFIN algorithm
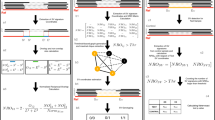
The underlying search algorithm of SMUFIN comprises two major steps (Fig. 1). First, under the assumption that any somatic variation occurring in the tumor genome will generate a unique sequence, tumor-specific reads are identified and isolated. This is achieved by creating a quaternary sequence tree (implemented as a generalized suffix array) using all tumor and normal reads (Fig. 1). In this tree, genomic regions of unaltered sequence will generate identical tumor and normal reads, and these will cluster together in common branches. Reads covering sequence variations in one or both alleles of the tumor are expected to form isolated branches without normal reads. These unique reads are then grouped into read blocks, each expected to cover a single sequence change or break in the tumor. By further interrogating the tree for overlapping regions (of at least 30 bp), each of these blocks is further expanded by adding and aligning the corresponding normal reads.

(a) (Left) As input, SMUFIN takes high-quality read data (FASTQ) of normal and tumor genomes of the same individual. (Middle) Starting and ending nucleotide sequences of representative example reads from tumor and normal samples. Reads containing no somatic mutations are shown in blue. Somatic mutations and downstream sequences are red. Nucleotide positions are indicated at the bottom, where n corresponds to the size of the read (Supplementary Fig. 4). Reads are numbered on the right side of the boxes. Pairs 1 and 1′, 3 and 3′, and 6 and 6′ would cover the same region in the nonmutated and mutated allele of the cancer genome, respectively. The other reads represent the two nonmutated alleles. (Right) These reads have different properties inside the quaternary tree. Because nonmutated cancer reads are expected to have their counterpart among healthy reads, they are also expected to share the same branches. Cancer reads that carry variations are expected to be unique and, therefore, to be located in isolated branches. These branches become cancer-specific exactly at the point where they differ, that is, in a breakpoint. SV, structural variation. (b) SMUFIN collects all the reads expanding on these cancer-specific branches and takes them as reads containing potential somatic variant breakpoints. Because any particular breakpoint is expected to be represented by several reads, we group all detectable reads that are overlapping and complementary and construct breakpoint blocks (Supplementary Fig. 5), covered by only one (single orientation) or by two strands (double orientation). This step, which includes filters for minimum overlap and coverage, removes a large fraction of false-positive variations, mostly derived from sequencing errors. (c) Each of the accepted blocks is then analyzed, as to the type of change detected. First, small variants, which can be defined within a single block, are identified. These include SNVs and small insertions, deletions and inversions. The remaining unclassified blocks are then passed into the next step where sequence translocations of large structural variants are defined. Here, for each of the breakpoints, we interrogated the tree and retrieved up to 100 bp of overlapping normal and tumor reads at each side of the break. (d) Finally, small and large variants are unambiguously positioned onto the reference genome by mapping18 the normal consensus region covering and flanking each of the variants. BWA, Burrows-Wheeler Aligner.
Next, potential tumor variants are defined and classified on each of the breakpoint blocks in two steps (Fig. 1). First, 'small' variants are identified—that is, SNVs and structural variants that can be completely defined within the size of a read. Second, 'large' structural rearrangements, which expand beyond the size of the input read, are defined. We expect that each of these blocks will represent one of the breaks generated by large insertions, inversions or deletions in the tumor genome, or to single translocation points. SMUFIN provides to the user these large structural variants as single breakpoints along with the corresponding surrounding sequence in the tumor. A simple filtering scheme is also used to ensure a minimum of physical coverage of all detectable variants and to correct for potential contamination of tumor cells in normal samples. Although default parameters have been adjusted in SMUFIN for common sequencing scenarios (i.e., ≥30-fold coverage depth in Illumina sequencing platforms), the user can also tune these filters to adapt the method to the particular characteristics of the data.
In summary, distinct features of SMUFIN that are not available in existing strategies for the detection of somatic variants include (i) the direct comparison of normal and tumor reads without the need to generate mapped BAM files; (ii) the detection, in a single execution, of SNVs and structural variants, such as inter- and intrachromosomal translocations, inversions, insertions and deletions of any size; (iii) the identification of variants at base pair resolution; and (iv) the reconstruction of exact changes in the tumor genome, including the sequence at both sides of all breakpoints detected.
Furthermore, we have developed a Message Passing Interface (MPI) implementation of SMUFIN that yields direct improvements of its usability and execution times. Using 16 nodes (2xIntel SandyBridge, 8-core/2.6 GHz) SMUFIN was able to complete the analysis of a tumor-normal, whole-genome pair in 4–8 h for samples with 30× of sequencing coverage, and 9–15 h for 60× samples. These executions showed discrete peaks of RAM usage of 8–10 Gb and 13–17 Gb per node, respectively.
Assessment and comparison of SMUFIN with model genomes
To assess SMUFIN's performance, we measured both the fraction of somatic variants detected (sensitivity) and the precision of this detection (specificity) using simulated and real cancer genome data together with orthogonal experimental techniques.
We generated normal and tumor test genomes by first applying to the human reference genome the sequence variation corresponding to a random human haplotype9 and to a predesigned catalog of somatic changes, and then simulating whole-genome sequencing at different depths of coverage (Online Methods, Supplementary Fig. 1 and Supplementary Table 1). To assess the applicability of SMUFIN in the current context of cancer genome analysis, we compared its performance with a representative set of somatic variant callers that are common parts of current pipelines for the analysis of tumor genomes: Mutect for SNVs3, and BreakDancer6, Pindel10, Delly7 and CREST11 for structural variants of different sizes (Supplementary Table 2). For the present comparison, we ran them as described in their companies' corresponding publication or website.
We first observed that the calling of somatic SNVs was nearly optimal and within the same range in Mutect and SMUFIN, with sensitivities of 97% and 92%, and specificities of 93% and 99%, respectively (Table 1 and Supplementary Table 3). On the other hand, the calling efficiency of somatic structural variants varied greatly between different methods, revealing clear differences when compared to SMUFIN. Some methods reached reasonable levels of sensitivity when the evaluation was restricted to the range of structural variants they were designed to detect (Pindel and Delly), but these dropped drastically when compared against the complete catalog of structural variations in the tumor (Supplementary Table 4). By contrast, SMUFIN was able to identify somatic structural variants with a sensitivity of 74% independently of the size of the structural variant, reaching >90% sensitivity when only structural variants larger than the read size were taken into account. SMUFIN's sensitivity for somatic SNV and structural variant calling is actually similar to that resulting from the combination of all the methods above: 94% versus 89% for SMUFIN.
The downside of combining these methods as a strategy for variant calling is the low levels of specificity achieved. In fact, in terms of specificity, the values for the external structural variant callers were 29–77%, whereas SMUFIN reached values of 91% across all structural variants. We also tested for consistency at sensitivity level in the identification of medium structural variants (i.e., variant size of 5–500 bp), which constitute a group of variants that have been particularly challenging for structural variant-calling methods that rely on pre-aligned data. This analysis showed that only SMUFIN and Pindel, which has been specifically designed also for small structural variants, kept a similar sensitivity when compared with the identification of the total of structural variants (Supplementary Table 4). When further testing SMUFIN, Pindel and CREST using lower levels of in silico sequencing coverage, we observed an overall decrease in performance, both at sensitivity and specificity levels, at physical sequencing coverage below 20-fold (Supplementary Fig. 2).
Detection of small somatic variants in human tumors
To further investigate the performance of SMUFIN in real data, we calculated and assessed the positive discovery rate of somatic SNVs and structural variants calling using whole-genome sequence (WGS) data from primary tumor and matched nontumor samples. We first tested the detection of small variants by analyzing a previously described sample (M004) of mantle cell lymphoma (MCL)12, an aggressive subtype of lymphoid neoplasia. SMUFIN identified 4,409 somatic SNVs and 1,094 small structural variants (Supplementary Table 5). To evaluate the specificity of SMUFIN, we verified >94% of SNVs (76 of 81) and >80% of structural variants (28 of 35) from a random set of 111 of these somatic calls by Sanger sequencing using the same DNA used for whole genome sequencing (Supplementary Table 6). These specificity rates are in agreement with the corresponding values obtained from the in silico analysis.
Complex structural variation in aggressive tumors
We next evaluated SMUFIN's accuracy in detecting large structural variants involving the somatic insertion, deletion, inversion or translocation of DNA fragments that are hundreds to millions of base pairs in length. For this test, we analyzed whole-genome sequence data from another mantle cell lymphoma sample (M003) and a sample from a pediatric form of a medulloblastoma (MB1), both known to present complex landscapes of chromosomal rearrangements12,13. Because these representative examples corresponded to a hematological and a solid tumor, each sequenced in a different sequencing facility, this analysis also measured SMUFIN's consistency across different types of data.
Identification of chromothripsis
MB1 was previously described as presenting chromothripsis, a complex structural alteration of the genome hypothesized to arise from a single catastrophic event that generates multiple breakpoints, often affecting one single chromosome14. In this tumor sample, SMUFIN uncovered a total of 102 breakpoints corresponding to large structural variants (i.e., beyond the read size), covering 85 intra- and 17 interchromosomal translocations (Supplementary Table 7). From the assessment of a random set of 39 of these breaks through PCR amplification and Sanger sequencing, we verified 36 (92%). Among all the breakpoints detected, 25 agreed with the intervals of chromosomal translocations that previously led to the definition of chromothripsis in this tumor, including three of the four verified at base-pair resolution.
In addition, we detected 65 previously unidentified breakpoints in the same tumor, covering 53 intra- and 12 interchromosomal translocations (Supplementary Fig. 3). From a random subset of 37 of these translocations (16 intra- and 11 interchromosomal), we verified 25 (92.5%) using Sanger sequencing. Together with the clusters of breakpoints already reported for chromosomes three and four in this tumor, new calls uncovered by SMUFIN enabled us to define a third damaged region in chromosome 11, with a density of six DNA breaks per Mb (between positions 39 and 45 Mb). Notably, many of these breakpoints correspond to translocations with chromosome 17 (Fig. 2). Furthermore, and complementary to the previous functional characterization of this tumor, we identified affected genes that were not reported in the previous study (Supplementary Table 7), including some that have been identified as possible driver genes, such as NCOR-1, SIN3P, WDR52 and PALLD, in several types of tumors15. Of the 65 breakpoints, 54 were predicted (allowing up to 100-nt deviation in the prediction) by at least one of the methods used above for the comparative assessment of SMUFIN, with 44 found only by Delly. This is not surprising considering the results of the in silico analysis, as sensitivity is not the major limitation of the reference alignment-dependent approaches.

(a) Circos representation of a genome-wide view of all the intra- and interchromosomal translocations identified by SMUFIN in this tumor (chromosomes with no breakpoints are excluded). Novel breakpoints are displayed in red, whereas those already reported are in gray. Breaks marked with “*” correspond to those that were tested and could be confirmed, resulting in a local specificity of 100%. Shadowed area indicates the interconnection between two regions in chromosomes 11 and 17 with high density of DNA breakage and rejoining events. (b) Circos map displaying all the breakpoints of chromosome 11 (within the 37–45 Mb region) and the interaction with chromosome 17 (15–19 Mb) in more detail. Genes affected by, at least one previously undescribed breakpoint are drawn, along with the exact position of the break.
Identification of chromoplexy
We also analyzed a sample from an aggressive form of mantle cell lymphoma (M003), previously described to have undergone complex chromosomal rearrangements12. We used SMUFIN to identify 30 breakpoints corresponding to large structural variants (Supplementary Table 8). Using PCR amplification followed by Sanger sequencing, we verified 19 of the 22 breakpoints tested, involving 7 intra- and 15 interchromosomal translocations (Supplementary Table 6). This not only confirms the correct location and the type of translocation identified, but it also shows that SMUFIN was able to reconstruct the correct sequence around the variants, as five of the breakpoints (six inter- and one intrachromosomal; Supplementary Table 8) included stretches of a new DNA insertion 5–30 nt long.
We next evaluated whether SMUFIN could be used to define the chromosomal arrangement of this tumor. We compared all 30 breakpoints identified, with 18 noncentromeric and nontelomeric regions of chromosomal imbalances previously detected using Affymetrix SNP6.0 array (Affymetrix, Santa Clara, CA)12. SMUFIN could redefine, at base pair resolution, 16 of these 18 regions. By manually assembling the fragments between all the translocations detected, we could model the landscape of this genome, which included three derivative chromosomes formed by combinations of large fragments of chromosomes 3 and 12 with smaller parts of chromosome 4. These chimeric chromosomes were experimentally confirmed in the mantle cell lymphoma cells by a combination of multicolor fluorescence in situ hybridization (FISH) and whole-chromosome painting analysis (Fig. 3). Furthermore, the resolution provided by SMUFIN allowed the identification of the fragmentation and fusion of genes not previously described in this sample. For example, we found that these translocations caused the fusion of ANK2 and SOX5 genes. Notably, these two rearrangement events did not appear to be independent as the corresponding fragments generated after the double-strand break were rejoined again reciprocally—that is, generating both, 12 to 4 and 4 to 12 translocations and two different forms of ANK2-SOX5 fusions (Fig. 3). In fact, 8 out of the 18 breakpoints appeared to be rejoined reciprocally, as recently described in prostate tumors16,17, suggesting an original organization of the chromatin where these regions were physically proximal and somehow interacting. A third translocation identified in the M003 tumor implies the breakage and putative inactivation of ARID2, a gene involved in chromatin remodeling.

(a) Three chimeric chromosomes formed by parts of chromosomes 3, 4 and 12 and the primary hallmark MCL translocation t(11;14). These rearrangements were identified by SMUFIN and all were experimentally verified by PCR. (b) A representative 24-color multicolor-FISH (mFISH) karyogram (top) that shows an unbalanced karyotype, with the t(11;14)(q13;q32) (BKP 10 and 11), a centromeric deletion of 17p, and several rearrangements between chromosomes 3, 4 and 12, all of them consistent with the breakpoints identified by SMUFIN. Bottom image shows a metaphase hybridized with whole-chromosome painting (WCP) 4 (green) and 12 (orange) probes showing four derivative chromosomes with material of these two chromosomes. Combination of mFISH and WCP analysis confirmed the presence of two different derivative chromosomes der(3)t(3;4;12), one der(12)t(3;4;12), and identified a fourth, der(4)t(4;12), which is not detectable by SMUFIN owing to the centromeric location of the breakpoint in chromosome 4. Scale bar, 10 μm. (c) Genes affected by chromoplexy—a reciprocal fusion of two genes (ANK2, in green and SOX5, in red) and a truncated chromatin remodeler (ARID2). Coding and noncoding exons are displayed as taller and shorter boxes, respectively.
By considering the number of rearrangements identified in this tumor, their distribution and the number of chromosomes involved, we classify this scenario as chromoplexy, a recently described phenomenon that, in contrast to chromothripsis14, is characterized by the presence of tens of unclustered chained rearrangements involving two or more chromosomes16,17. The high fraction of reciprocal rejoining events found in this tumor, together with the fusion of genes and the disruption of a chromatin remodeler gene, is also in agreement with the results of the chromoplectic events identified in prostate tumors.
Conclusions
We describe SMUFIN, a methodology for the identification of somatic variation in tumor genomes from their direct comparison with their corresponding normal samples. SMUFIN also provides an integrated solution for the identification, in a single run, of somatic SNVs and structural variants (insertions, deletions, inversions and translocations of any size), which can currently be partially achieved only by combining several independent programs and in-house filtering schemes into complex computational pipelines. Our method defines, at base pair resolution, complex scenarios of chromosomal rearrangements, such as chromoplexy and chromothripsis. SMUFIN was able to identify the translocations defined before using other computational and experimental methods, as well as novel breakpoints that complete the corresponding landscapes of chromosomal rearrangements. Owing to the underlying mechanism of the algorithm used in SMUFIN, our method is not suitable to quantify copy number variations or detect complete losses of chromosome arms or inversions flanked by palindromic sequences.
Beyond the benefits of the detection capabilities of SMUFIN, the current parallel implementation of the program also shows substantial improvements at the level of usability and execution time compared with available pipelines, as it can currently analyze a pair of whole genome sequences with coverage of 30–60× in 4–15 h, using 50–80 standard cores and requiring less than 17 Gb of RAM memory per computing node. This, together with the scalability of the program, will realistically allow a systematic and parallel analysis of cancer samples, accessible to nonexpert users with standard computing resources.
Taken together, the underlying search mechanism of SMUFIN constitutes an alternative way of processing and analyzing genomic data, which can inspire the development of new tools for other types of genomic analyses. Because SMUFIN actually finds changes in one sequence set relative to another, it could potentially be adjusted to other types of biomedical and evolutionary studies that rely on the comparative analysis of two genomes, even if they are from different species.
Methods
The SMUFIN algorithm.
The general structure and the internal mechanism of SMUFIN is displayed in Figure 1. The complete variant identification and characterization process comprises the following specific steps:
Input data. As input, SMUFIN takes high-quality sequencing data directly from FASTQ files of tumor and normal samples of the same individual. Alternatively, SMUFIN is also able to accept BAM files, from which it extracts all the sequencing reads. Sequences having over 10% of its bases with a phred quality score < q20 are discarded.
Construction of the quaternary sequence tree. A 'quad-tree'-based structure is first generated using all high-quality normal and tumor reads. All these sequences are sequentially loaded into the tree on the basis of their sequence (Fig. 1 and in Supplementary Fig. 4a). Each node of the tree has, at most, four branches, each one representing one of the four nucleotides. To avoid sequence ambiguity derived from the complexity of the genome, only fragments of at least 30 bp are inserted into the tree. In the case of the presence of undefined base pairs (“N”), these are removed and the original sequence is split forming new shorter reads, which are inserted in the tree only if they are longer than 30 base pairs. Each of sequences accepted is inserted into the tree, from the root, in original form (i.e., starting from nucleotide 1 to the end of the read), together with all derived suffixes larger than 30 bp (recursively starting from nucleotide 2 to the end, 3 to the end, etc...; Supplementary Fig. 4a). Because posterior searches through the tree start from the root, the presence of read suffixes allows a rapid identification of particular sequences and reads.
Selecting reads containing candidate variants. Once all the sequences and derived suffixes are loaded into the tree, the next step consists in identifying all tumor-specific reads. Because we expect that variants generate new and distinct sequences in the mutated genome compared with the nonmutated sample, SMUFIN first searches and collects sequences (reads) that are only present in the tumor sample. These sequences are identified from the tree, as nodes and branches with an unbalanced representation of normal (count normal reads; CNR) and tumor (count tumor reads; CTR) reads (Supplementary Fig. 4b). We expect that nodes or branches covering a variation in the tumor sequence will theoretically have no representation of normal reads. To favor this condition, we start to search the tree from the level 30 toward the leafs. We accepted only nodes and branches that have a CTR of at least 4. Internal tests suggest that setting CTR ≥ 4 improves specificity in a factor 1.4× with a negligible loss of sensitivity (not shown). Additionally, nodes or branches with a CNR to CTR ratio below a certain threshold (E_CONT) are selected. This threshold can be adjusted by the user to account for expected levels of contamination of tumor cells into the normal sample. Please, be aware that an E_CONT of 0 implies no expected contamination, that is, no acceptance of reads coming from the normal sample (CNR) on that candidate variant node or branch, which implies lower final sensitivity but higher specificity. On the other hand, an E_CONT larger than 0 always results in a higher sensitivity, but at the cost of lower specificity. E_CONT was set to 0 for the in silico analysis and to 0.05 for the real tumor samples analyzed here, where we assume a maximum of 5% contamination of tumor reads into the normal sample.
Grouping candidate reads. After all detectable tumor-specific reads have been identified, the next step consists in grouping those that are suspected to cover the same variant. For this, candidate sequences are organized by identity: two sequences belong to the same group if they overlap by at least 30 bp. Reverse complementary sequences are also evaluated during this grouping in order to be able to cover the variant in both orientations. Sequence blocks (groups) with sequences in only one of the orientations or with less than four tumor reads are discarded. Once these groups are generated, we interrogate the tree, also on the 30-bp overlap basis, to extract the normal (nonmutated) reads of the same region and add them to the block. Ideally, each block will represent a region in the genome containing the mutated and the nonmutated version (see a detailed example of a breakpoint block in Supplementary Fig. 5). In order to classify and characterize the type of variation identified, we extract the consensus mutated and normal sequences from these blocks. Normal consensus sequences will be also used at the end of the procedure and mapped onto the reference genome to obtain the coordinates of the variant.
Identification and characterization of variants. Once all possible breakpoint blocks are defined, the next step consists in identifying and classifying the variation included there. Normal and tumor consensus sequences derived from these blocks (Supplementary Fig. 5) are recursively compared to identify differences. A first evaluation will search for small variants, which consist of those that are completely included within the consensus sequences (SNV and small structural variants: insertions, deletions and inversions). All the blocks that do not match this criterion are then considered candidates for large structural variants, that is, those likely to cover breakpoints of intra- or interchromosomal transitions, part of large deletions, insertions, inversions or translocations. In this case, each tumor consensus sequence is extended on both ends (Fig. 1) by interrogating the tree for unambiguous tumor reads that overlap at least 30 bp with the tumor consensus, reconstructing a (maximum) 200-bp region around the break and allowing the detection of newly generated sequence at the point of the break.
After small and large somatic variants are defined, we identify the coordinates of the changes by mapping onto the reference genome the normal consensus sequences corresponding to each of the variants, avoiding potential mapping conflicts derived from the presence of the variant, as usually happens when using reference-based approaches. Sequences mapping (with the same score) to several positions in the genome are discarded.
Calibration and default parameters for SMUFIN were adjusted using a high-quality set of ∼1,000 SNVs identified with the Sidrón software in a chronic lymphocytic leukemia sample4.
SMUFIN's pseudo-code.
SeqReader normalReader = openSeqReader(normal_input_file); SeqReader tumorReader = openSeqReader(tumor_input_file); Tree qtree = initTree(); Foreach read in normalReader: If quality_check(read): insertIntoTree(qtree, read, as_normal); Foreach read in tumorReader: If quality_check(read): insertIntoTree(qtree, read, as_tumor); List candidate_reads = GenerateEmptyList(); Foreach node in qtree: If depth(node) >= 30 and CTR(node) >= 4 and CNR(node)/CTR(node) < E_CONT): reads = GetTumorReadsFromNode(node); InsertReadsIntoList(candidate_reads, reads); List breakpoint_blocks = GenerateEmptyList(); Foreach read in candidate_reads: tumor_reads = GetOverlappingReadsFromCandidateReads(read, candidate_reads); normal_reads = GetOverlappingReadsFromTree(tumor_reads, qtree, as_normal); bp_block = GenerateBPBlock(normal_reads, tumor_reads); If Coverage(bp_block) >= 4: InsertIntoList(breakpoint_blocks, bp_block); List large_variant_candidates = GenerateEmptyList(); Foreach bp_block in breakpoint_blocks normal_consensus_sequence = GetNormalConsensusSequenceFromBPBlock(bp_block); tumor_consensus_sequence = GetTumorConsensusSequenceFromBPBlock(bp_block); If HasSmallVariant(normal_consensus_sequence, tumor_consensus_sequence) align_info = MapSequenceToReference(normal_consensus_sequence) If (UnambiguousMapping(align_info) outputSmallSV(align_info, bp_block); Else InsertIntoList(large_variant_candidates, bp_block); Foreach bp_block in large_variant_candidates extended_sequence = ExtendTumorSequenceFromBPBlock(bp_block, qtree); align_info = MapExtendedToReference(extended_sequence); If UnambiguousMapping(align_info) OutputLargeSV(align_info, extended_sequence);
Construction of the in silico genome.
A personalized genome was simulated using the hg19 reference genome downloaded from UCSC (with no repeat-masking), and modifying it to match a randomly chosen human haplotype from the 1000 Genome database. These 7,194,026 variants consist of 4,745,917 SNPs and 2,447,367 deletions. The complete list of these germline events can be found at http://cg.bsc.es/smufin/download. The catalog of somatic variants further added to this personalized genome includes 8,240 SNVs (more than 100 bp apart), 20 known tumor translocations19,20, 715 random insertions, 738 random deletions and 345 random inversions, all ranging from 1 bp to 100 Mbp (Supplementary Fig. 4 and Supplementary Table 1). In silico sequencing was simulated using ART Illumina21. For this, we first generated a profile using the M004 sample to extract parameters, like sequence variation or read length. We then run the program at different depths of coverage, using the resulting parameters and a default error rate (0.00009).
Analysis of the in silico genome with external methods.
Each of the external methods for the comparison with SMUFIN was run on pooled libraries (normal and tumoral) using default settings except for the following parameters: BreakDancer was run with -q 10 (mapping quality) and score cutoff of >80, as described before6,22; Pindel's results with less than five supporting reads were not considered as recommended elsewhere to increase specificity23; predictions obtained with Delly were rejected if the number of supporting reads were less than three and the mapping quality 20. For BreakDancer, Pindel and Delly, somatic variants were obtained by filtering out all the structural variants found in both normal and tumor libraries: we only kept those structural variants with no unique supporting reads from the normal library. CREST and Mutect already provided somatic variants as direct results. BreakDancer and Pindel were used as complementary methods covering large and small structural variants, respectively, as advised by the developers.
Data sets.
M003, M004 and MB1 were obtained with informed consent and an ethical vote (Institutional Review Board) following ICGC guidelines (https://icgc.org). M003, M004 and MB1 were accessed through the European Genome-phenome Archive (EGA, https://www.ebi.ac.uk/ega/) under access numbers EGAS00001000510 and EGAS00001000085.
Identification and analysis of variant genes.
Variants genes in tumor samples were identified by analyzing all the changes identified with ANNOVAR24. The analysis of the resulting genes potentially modified at coding or splicing level were further analyzed with Intogen15 in order to infer their potential role in oncogenesis.
Experimental verification of variants.
PCR primers were designed on sequence blocks of 2,000 bp around the target variant using Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/)25. PCR reactions were performed for tumor and control samples. Each target locus was amplified using 50 ng of DNA. The amplification was performed using Qiagen Multiplex PCR Kit (Qiagen), and the reaction mix contained 2× QIAGEN Multiplex PCR Master Mix, 10× primer mix (2 μM of each primer) and RNase-free water until a total reaction volume of 25 μl. PCR conditions were as follows: 96 °C, 10 min; 2 cycles of 96 °C, 30 s/60 °C, 30 s/72 °C, 1 min 30 s; 2 cycles of 96 °C, 30 s/58 °C, 30 s/72 °C, 1 min 30 s; 2 cycles of 96 °C, 30 s/56 °C, 30 s/72 °C, 1 min 30 s; 35 cycles of 96 °C, 30 s/54 °C, 30 s/72 °C, 1 min 30 s/70 °C, 10 min. All the PCR products were run in a capillary electrophoresis gel (QIAxcel Advanced System, Qiagen) with the QIAxcel DNA screening kit (Qiagen), and the multiband PCR products were purified using NucleoSpin Gel and PCR Clean-up (Mercherey-Nagel). Regarding the Sanger sequencing, PCR products were cleaned using ExoSAP-IT (USB) and sequenced using ABI Prism BigDye terminator v3.1 (Applied Biosystems) with 5 pmol of each primer. Sequencing reactions were run on an ABI-3730 Sanger sequencing platform (Applied Biosystems). Sequences were examined with the Mutation Surveyor DNA Variant Analysis Software (Softgenetics).
G-banding, FISH and M-FISH analysis.
Conventional cytogenetics was performed on Giemsa-banded chromosomes (G-banding) obtained after a 72-h culture and stimulation with tetradecanoyl-phorbol-acetate. Results of the ten metaphases analyzed were described according to the International System for Human Cytogenetic Nomenclature26. FISH studies for the presence of the t(11;14) translocation and 17p deletions were performed using Vysis LSI IGH/CCND1 Dual Color Dual Fusion and Vysis LSI TP53 (17p13.1) (Abbott Molecular, Des Plaines, IL) on fixed cells according to the manufacturer's specifications. Two hundred nuclei were examined for each probe. To identify the chromosomes involved in marker chromosomes and to disclose other possible structural balanced abnormalities, we performed 24-color karyotyping using 24XCyte human multicolor FISH (mFISH) probe kit according to manufacturer's instructions (MetaSystems, Altlussheim, Germany) consisting of 24 different chromosome painting probes (combinatorial labeling). Image capture was done with Nikon Eclipse 50i equipped with a CCD-camera (CoolCube1, MetaSystems) and appropriate filters using Isis software. Karyotyping was done using the 24-color mFISH upgrade package. Additionally, whole chromosomal paintings (WCP) of chromosome 4 (spectrum green) and 12 (spectrum orange) were performed simultaneously.
Figure 3 was done using CIRCOS software27.
Accession codes.
For validation sequences produced in this study (Supplementary Table 6), European Genome-phenome Archive: EGAS00001000510.
References
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
Frampton, G.M. et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31, 1023–1031 (2013).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Puente, X.S. et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 475, 101–105 (2011).
Degner, J.F. et al. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics 25, 3207–3212 (2009).
Chen, K. et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681 (2009).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Alexandrov, L.B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Ye, K., Schulz, M.H., Long, Q., Apweiler, R. & Ning, Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871 (2009).
Wang, J. et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods 8, 652–654 (2011).
Beá, S. et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 110, 18250–18255 (2013).
Rausch, T. et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148, 59–71 (2012).
Korbel, J.O. & Campbell, P.J. Criteria for inference of chromothripsis in cancer genomes. Cell 152, 1226–1236 (2013).
Gonzalez-Perez, A. et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 10, 1081–1082 (2013).
Baca, S.C. et al. Punctuated evolution of prostate cancer genomes. Cell 153, 666–677 (2013).
Shen, M.M. Chromoplexy: a new category of complex rearrangements in the cancer genome. Cancer Cell 23, 567–569 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Richter, J. et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 44, 1316–1320 (2012).
Teles Alves, I. et al. Next-generation sequencing reveals novel rare fusion events with functional implication in prostate cancer. Oncogene 10.1038/onc.2013.591 (3 February 2014).
Huang, W., Li, L., Myers, J.R. & Marth, G.T. ART: a next-generation sequencing read simulator. Bioinformatics 28, 593–594 (2012).
Young, M.A. et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell 10, 570–582 (2012).
Jones, D.T. et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 45, 927–932 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Untergasser, A. et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
Shaffer, L.G., McGowan-Jordan, J. & Schmid, M. (eds.) ISCN 2013: An International System for Human Cytogenetic Nomenclature (2013) (Kargar, 2013).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Acknowledgements
The ICGC-CLL Genome Consortium is funded by the Spanish Ministry of Economy and Competitiveness (MINECO) through the Instituto de Salud Carlos III (ISCIII), Red Temática de Investigación del Cáncer (RTICC) of the ISCIII (RD12/0036/0036) and National Institute of Bioinformatics (INB). This study was also supported by Ministerio de Economía y Competitividad, Secretaría De Estado De Investigación, Desarrollo e Innovación PLAN NACIONAL de I+D+i 2008-2011, Subprograma de Apoyo a Centros y Unidades de excelencia Severo Ochoa; and Plan Nacional SAF12/38432; Generalitat de Catalunya AGAUR 2009-SGR-992; Fondo de Investigaciones Sanitarias (PI11/01177); Association for International Cancer Research (12-0142). J.O.K. and M.S.W. were supported by the European Commission (Health-F2-2010-260791). C.L.-O. is an investigator of the Botin Foundation. E.C. and M.O. are ICREA Academia Researchers. We also thank S. Guijarro and C. Gómez for their excellent technical assistance.
Author information
Authors and Affiliations
Contributions
V.M., S.G. and D.T. conceived and designed the study. L.O.A., L.M., M.P., J.L.G., R.R. and M.O. performed data analysis. S.B., I.S., C.R., A.N., E.C. and I.G.G. generated and experimentally validated the MCL samples. M.S.-W., A.M.S. and J.O.K. generated and experimentally validated the MB1 sample. C.L.-O., X.S.P., E.C. and D.T. wrote the manuscript; and D.T. supervised the whole study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5 and Supplementary Tables 2 and 4 (PDF 4689 kb)
Supplementary Table 1 (XLSX)
List of somatic modifications affecting the in silico genome (XLSX 951 kb)
Supplementary Table 3 (XLSX)
Results of SMUFIN and others methods for the in silico validation analysis (XLSX 5687 kb)
Supplementary Table 5 (XLSX)
Results of SMUFIN on the Mantle Lymphoma M004 (XLSX 773 kb)
Supplementary Table 6 (XLSX)
Validation sequences of M003 lage structural variants. The identifyiers shown here correspond to the ones of Supplementary Table 8. (XLSX 23 kb)
Supplementary Table 7 (XLSX)
Results of SMUFIN on the Medulloblastoma MB1 (XLSX 208 kb)
Supplementary Table 8
Results of SMUFIN on the Mantle Lymphoma M003 (XLSX 194 kb)
Supplementary Source Code
Compressed ZIP file with SMUFIN's source code (ZIP 1659 kb)
Rights and permissions
About this article

Cite this article
Moncunill, V., Gonzalez, S., Beà, S. et al. Comprehensive characterization of complex structural variations in cancer by directly comparing genome sequence reads. Nat Biotechnol 32, 1106–1112 (2014). https://doi.org/10.1038/nbt.3027
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.3027
This article is cited by
-
Ultrafast prediction of somatic structural variations by filtering out reads matched to pan-genome k-mer sets
Nature Biomedical Engineering (2022)
-
GeDi: applying suffix arrays to increase the repertoire of detectable SNVs in tumour genomes
BMC Bioinformatics (2020)
-
Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma
Nature Genetics (2020)
-
Retrospective evaluation of whole exome and genome mutation calls in 746 cancer samples
Nature Communications (2020)
-
Pan-cancer analysis of whole genomes
Nature (2020)
