
Abstract
Plant-parasitic nematodes are major agricultural pests worldwide and novel approaches to control them are sorely needed. We report the draft genome sequence of the root-knot nematode Meloidogyne incognita, a biotrophic parasite of many crops, including tomato, cotton and coffee. Most of the assembled sequence of this asexually reproducing nematode, totaling 86 Mb, exists in pairs of homologous but divergent segments. This suggests that ancient allelic regions in M. incognita are evolving toward effective haploidy, permitting new mechanisms of adaptation. The number and diversity of plant cell wall–degrading enzymes in M. incognita is unprecedented in any animal for which a genome sequence is available, and may derive from multiple horizontal gene transfers from bacterial sources. Our results provide insights into the adaptations required by metazoans to successfully parasitize immunocompetent plants, and open the way for discovering new antiparasitic strategies.
Similar content being viewed by others


Main
Plant-parasitic nematodes are responsible for global agricultural losses amounting to an estimated $157 billion annually. Although chemical nematicides are the most reliable means of controlling root-knot nematodes, they are increasingly being withdrawn owing to their toxicity to humans and the environment. Novel and specific targets are thus needed to develop new strategies against these pests.
The Southern root-knot nematode Meloidogyne incognita is able to infect the roots of almost all cultivated plants, making it perhaps the most damaging of all crop pathogens1. M. incognita is an obligatory sedentary parasite that reproduces by mitotic parthenogenesis2. Root-knot nematodes have an intimate interaction with their hosts. Within the host root, adult females induce the redifferentiation of root cells into specialized 'giant' cells, upon which they feed continuously (Fig. 1). M. incognita can infect Arabidopsis thaliana, making this nematode a key model system for the understanding of metazoan adaptations to plant parasitism3,4 (Supplementary Data, section 1 online).

Infective second-stage juveniles (J2) penetrate the root and migrate between cells to reach the plant vascular cylinder. The stylet (arrowhead) connected to the esophagus is used to pierce plant cell walls, to release esophageal secretions and to take up nutrients. Each J2 induces the dedifferentiation of five to seven root cells into multinucleate and hypertrophied feeding cells (*). These giant cells supply nutrients to the nematode (N). The nematode becomes sedentary and goes through three molts (J3, J4, adult). Occasionally, males develop and migrate out of the roots. However, it is believed that they play no role in reproduction. The pear-shaped female produces eggs that are released on the root surface. Embryogenesis within the egg is followed by the first molt, generating second-stage juveniles (J2). Scale bars, 50 μm.
The phylum Nematoda comprises >25,000 described species, many of which are parasites of animals or plants2. As many as 10 million species may have yet to be described. Although the model free-living nematodes Caenorhabditis elegans and Caenorhabditis briggsae have been the subjects of intensive study5,6, little is known about the other members of this diverse phylum. These two free-living models will likely not illuminate the biology of nematode parasitism (Supplementary Fig. 1 online), as shown by the substantial differences between their genome sequences and that of the human parasite Brugia malayi7.
The genome sequence of M. incognita presented here provides insights into the adaptations required by metazoans to successfully parasitize and counter defenses of immunocompetent plants, and suggests new antiparasitic strategies.
Results
General features of the M. incognita genome
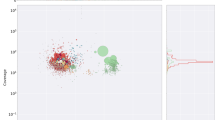
The M. incognita genome was sequenced using whole-genome shotgun strategy. Assembly with Arachne8 yielded 2,817 supercontigs, totaling 86 Mb (Table 1; Supplementary Data, section 2; Supplementary Fig. 2; Supplementary Table 1 online)—almost twice the estimated genome size (47- to 51-Mb haploid genome)9. All-against-all comparison of supercontigs revealed that 648 of the longest (covering ∼55 Mb) consist of homologous but diverged segment pairs (Fig. 2) that might represent former alleles (Supplementary Data, section 2; Supplementary Figs. 3 and 4 online). About 3.35 Mb of the assembly constitutes a third partial copy aligning with these supercontig pairs. Average sequence divergence between the aligned regions is ∼8% (Fig. 3). A combination of different processes may explain the observed pattern in M. incognita, including polyploidy, polysomy, aneuploidy and hybridization10,11; all are frequently associated with asexual reproduction. These observations are consistent with a strictly mitotic parthenogenetic reproductive mode, which can permit homologous chromosomes to diverge considerably, as hypothesized for bdelloid rotifers12 (Supplementary Data, section 2.2). No DNA attributable to bacterial endosymbiont genome(s) was identified.

The five largest supercontigs are shown with plots of gene density (orange curve), conservation with C. elegans at amino acid level (green curve) and EST density (pink curve). Blue lines represent most similar matches at the protein level between each predicted gene on these five supercontigs and 70 matching supercontigs.

Exons are represented by red boxes and are linked together to form genes (arrows indicate the direction of transcription). Gray boxes show assembly gaps. Highly diverged allelic genes are linked together using blue boxes. Gene order is well conserved between the two allelic-like regions, with only minor differences in predicted gene structure. Percentages of sequence identity at the protein level between the two allelic-like regions are indicated.
Noncoding DNA repeats and transposable elements represent 36% of the M. incognita genome (Supplementary Data, section 3; Supplementary Figs. 5 and 6 and Supplementary Tables 2 and 3 online). One repeat family with 283 members on 46 contigs encoded the nematode trans-spliced leader (SL) exon, SL1, of which 258 members were found associated with a satellite DNA13 (Supplementary Fig. 7 online). In nematodes, many mature mRNAs share this 5′ SL exon, and trans-splicing is also associated with resolution of polycistronic pre-mRNAs derived from operons. We identified 1,585 candidate M. incognita operons containing a total of 3,966 genes. The two longest operons contained ten genes each and are not allelic copies (Supplementary Table 4 online). Operons are a dynamic component of nematode genome architecture, as different sets of genes were operonic in M. incognita, C. elegans and B. malayi, and only one operon was found to be strictly conserved between the three nematodes (Supplementary Data, section 4; Supplementary Figs. 8 and 9; Supplementary Table 5 online).
The gene content of a plant-parasitic nematode
The genome sequence was annotated using the integrative gene prediction platform EuGene14, specifically trained for M. incognita (Supplementary Data, section 5; Supplementary Table 6 online). We identified 19,212 protein-coding genes (Table 1). Due to the high variation between allelic-like copies (Fig. 3) potentially allowing functional divergence, all copies were considered to be different genes. Indeed, 69% of protein sequences were <95% identical to any other (Supplementary Table 7 and Supplementary Fig. 10 online). The protein-coding genes occupy 25.3% of the sequence at an average density of 223 genes Mb−1, and 36% are supported by expressed sequence tags (ESTs). InterPro protein domains were identified in 55% of proteins and 22% were predicted to be secreted. Comparison of domain occurrence in M. incognita with that in C. elegans identified an increased abundance of 'pectate lyase', glycoside hydrolase family GH5 and peptidase C48 (SUMO) domains, and fewer chemoreceptor domains. We compared the domain content of the M. incognita protein set to those of C. elegans, B. malayi, Drosophila melanogaster and three fungi, of which two are plant pathogens. Thirty-two domains were detected only in M. incognita, and two additional domains were only shared between the two plant-pathogenic fungi and M. incognita. Functions assigned to the 34 domains specific to plant pathogens encompassed plant cell-wall degradation and chorismate mutase activity (see below). OrthoMCL15 clustering of the same eight proteomes suggested that 52% of M. incognita predicted proteins had no ortholog in the other species. Among them, 1,819 proteins (of which 338 were supported by ESTs) are secreted and lack any known domain (Supplementary Data, section 6; Supplementary Figs. 11 and 12; Supplementary Tables 8, 9, 10 online). The core complement of proteins in the phylum Nematoda is relatively small: ∼23% of the ortholog groups were shared by M. incognita, C. elegans and B. malayi (Supplementary Fig. 12b).
Identifying plant parasitism genes
Nematode proteins produced in and secreted from specialized gland cells into the host are likely to be important effectors of plant parasitism4,16. We identified gene products that might be involved in parasitic interaction, particularly those that might modify plant cell walls.
M. incognita has an unprecedented set of 61 plant cell wall–degrading, carbohydrate-active enzymes (CAZymes). Although a few such individual CAZymes had been identified previously in some plant-parasitic nematodes and in two insect species4,16,17, they are absent from all other metazoans studied to date (Table 2; Supplementary Data, section 7.1; Supplementary Tables 11, 12, 13, 14 online). We identified 21 cellulases and six xylanases from family GH5, two polygalacturonases from family GH28 and 30 pectate lyases from family PL3. We also identified CAZymes not previously reported from metazoans, including two additional plant cell wall–degrading arabinases (family GH43) and two invertases (family GH32). Invertases catalyze the conversion of sucrose (an abundant disaccharide in plants) into glucose and fructose, which can be used by M. incognita as a carbon source. We also identified a total of 20 candidate expansins in M. incognita, which may disrupt noncovalent bonds in plant cell walls, making the components more accessible to plant cell wall–degrading enzymes18. This suite of plant cell wall–degrading CAZymes, expansins and associated invertases was probably acquired by horizontal gene transfer (HGT), as the most similar proteins (outside plant-parasitic nematodes) were bacterial homologs (Supplementary Table 12). M. incognita also has four secreted chorismate mutases19, which most closely resemble bacterial enzymes. Chorismate mutase is a key enzyme in biosynthesis of aromatic amino acids and related products, and M. incognita may subvert host tyrosine-dependant lignification or defense responses. Overall, these genes suggest a critical role of HGT events in the evolution of plant parasitism within root-knot nematodes.
Apart from genes restricted to M. incognita, we also identified gene families showing substantial expansion compared to C. elegans. Among the most notable idiosyncrasies in M. incognita, we identified more than 20 cysteine proteases of the C48 SUMO (small ubiquitin-like modifier) deconjugating enzyme family—four times the number in C. elegans (Supplementary Data, section 7.2; Supplementary Table 15 online). As some phytopathogenic bacterial virulence factors are SUMO proteases20, the proteolysis of sumoylated host substrates may be a general strategy used by pathogens to manipulate host plant signal transduction. The M. incognita genome also encodes nine serine proteases from the S16 sub-family (Lon proteases), whereas only three are identified in C. elegans. These proteases regulate type III protein secretion in phytopathogenic bacteria21 and may have analogous roles in M. incognita.
We identified orthologs to other known candidate plant-parasitic nematode parasitism genes in the genome of M. incognita. As most of these gene families are also present in animal-parasitic nematodes and C. elegans, M. incognita members putatively involved in parasitism were probably recruited from ancestral nematode families (Supplementary Data, section 7.3; Supplementary Table 16 online). Twenty-seven previously described M. incognita–restricted pioneer genes expressed in esophageal glands22 were retrieved in the genome. Eleven additional copies were identified; all remain Meloidogyne spp. specific (Supplementary Data, section 7.4; Supplementary Table 17 online). These secreted proteins of as-yet-unknown function are likely targets for novel intervention strategies, and warrant deeper investigation.
Protection against environmental stresses
One aspect of plant defense responses is the production of cytotoxic oxygen radicals. However, M. incognita has fewer genes encoding superoxide dismutases and glutathione peroxidases than C. elegans (Supplementary Data, section 7.5; Supplementary Table 18 online). More striking still was the reduction in glutathione S-transferases (GSTs) and cytochromes P450 (CYPs), enzymes involved in xenobiotic metabolism and protection against peroxidative damage. Whereas C. elegans has 44 GSTs, including representatives from the Omega, Sigma and Zeta classes23, M. incognita possesses only 5 GSTs, all from the Sigma class. Sigma class GSTs are involved in protection against oxidants rather than xenobiotics. A comparable reduction in gst genes was observed in B. malayi7. Similarly, whereas C. elegans has 80 different cyp genes from 16 families24, only 27 full or partial cyp genes, from 8 families, were identified in M. incognita. CYP35 and other families of xenobiotic-metabolizing P450s are absent from M. incognita (Supplementary Data, section 7.5; Supplementary Table 18).
We identified M. incognita orthologs of all genes of the innate immunity signaling pathways of C. elegans25 except trf-1, which is part of the Toll pathway (Supplementary Data, section 7.5; Supplementary Table 19 online). However, immune effectors such as lysozymes, C-type lectins and chitinases were much less abundant in M. incognita than in C. elegans. As previously observed in B. malayi7, entire classes of immune effectors known from C. elegans were absent from M. incognita, including antibacterial genes such as abf and spp26 and antifungal genes of several classes (nlp, cnc, fip, fipr)25 (Supplementary Data, section 7.5; Supplementary Table 19). As plant parasites embedded in root tissues are protected from a variety of biotic and abiotic stresses, we speculate that the reduction and specialization of chemical and immune defense genes is a result of life in this privileged environment.
C. elegans has a broad range of unusual fucosylated N-glycan structures compared to other metazoans27. M. incognita has almost twice as many candidate fucosyltransferases as C. elegans (Supplementary Data, section 7.1; Supplementary Table 14). As suggested for animal-parasitic nematodes, multi-fucosylated structures on the surface of the nematode cuticle could help M. incognita to evade recognition27.
Core biological processes
Nuclear receptors, kinases, G-protein coupled receptors (GPCRs) and neuropeptides encompass some of the gene products most extensively involved in core physiological, developmental and regulatory processes.
C. elegans has a surprisingly large number of nuclear receptors, but curiously lacks orthologs of many nuclear receptor types conserved in other animals28. Some of these conserved nuclear receptors are present in B. malayi7. Among the 92 predicted nuclear receptors in M. incognita, we identified orthologs of several known nematode nuclear receptors, although many of the nuclear receptors present in B. malayi and absent in C. elegans were also absent in M. incognita (Supplementary Data, section 7.6; Supplementary Table 20 online). Many C. elegans nuclear receptors are classified as supplementary nuclear receptors (SupNRs), likely derived from a hepatocyte nuclear factor-4-like ancestor29. Orthologs of SupNRs were found in M. incognita, including a 41-member, M. incognita-specific expansion. Fourteen SupNRs are one-to-one orthologs between B. malayi, M. incognita and C. elegans, or conserved only between M. incognita and C. elegans, with secondary losses in B. malayi (Supplementary Data, section 7.6; Supplementary Fig. 13 online). Thus the expansion of SupNRs started before the Brugia-Meloidogyne-Caenorhabditis split and has proceeded independently in C. elegans and M. incognita.
M. incognita has 499 predicted kinases compared to 411 in C. elegans30 and 215 in B. malayi7. The kinases were grouped into 232 OrthoMCL clusters, 24 of which contained only nematode members, suggesting that they have nematode-specific functions. Four kinase families contained only M. incognita and B. malayi members, suggesting potential roles for these genes in parasitism. Finally, 66 kinase families, containing 122 genes, appear to be M. incognita-specific (Supplementary Data, section 7.7; Supplementary Table 21 online). Seven percent (1,280) of all C. elegans genes are predicted to encode GPCRs that play crucial roles in chemosensation. These C. elegans genes have been divided into three serpentine receptor superfamilies and five solo families31. M. incognita has only 108 GPCR genes and these derive from two of the three serpentine receptor superfamilies and one of the solo families. These M. incognita chemosensory genes are commonly found as duplicates clustered on the genome, as observed in C. elegans (Supplementary Data, section 7.8; Supplementary Fig. 14; Supplementary Table 22 online).
Neuropeptide diversity is remarkably high in nematodes, given the structural simplicity of their nervous systems. C. elegans has 28 Phe-Met-Arg-Phe-amide-like peptide (flp) and 35 neuropeptide-like protein (nlp) genes encoding ∼200 distinct neuropeptides32. The identified neuropeptide complement of M. incognita is smaller: 19 flp genes and 21 nlp genes. However, two flp genes, Mi-flp-30 and Mi-flp-31, encode neuropeptides that have not been identified in C. elegans, suggesting that they could fulfill functions specific to a phytoparasitic lifestyle (Supplementary Data, section 7.9; Supplementary Table 23 online).
The XX-XO sex determination pathway in C. elegans is intimately linked to the dosage compensation pathway33. M. incognita reproduces exclusively by mitotic parthenogenesis, and males do not contribute genetically to production of offspring11. M. incognita also displays an environmental influence on sex determination: under less favorable environmental conditions far more males are produced. These males can arise due to sex reversal34 and intersexual forms can be produced. M. incognita homologs of at least one member of each step of the C. elegans sex determination cascade were identified, including sdc-1 from the dosage compensation pathway, tra-1, tra-3 and fem-2 from the sex determination pathway itself, and also downstream genes such as mag-1 (which represses male-promoting genes) and mab-23 (which controls male differentiation and behavior). In addition, a large family (∼35 genes) of M. incognita secreted proteins, similar to the C2H2 zinc finger motif–containing tra-1 from C. elegans, was identified (Supplementary Data, section 7.10; Supplementary Table 24 online). It is therefore possible that M. incognita uses a similar genetic system for sex determination, but with the male pathway also modulated in response to environmental cues.
Taken together, these comparative analyses of genes, underpinning important traits, highlight the huge biodiversity in the phylum Nematoda. Idiosyncrasies identified in M. incognita may account for its parasitic lifestyle and lead to the development of new control strategies directed against plant-parasitic nematodes.
RNA interference and lethal phenotypes
RNA interference (RNAi) is a promising technology for the functional analysis of parasitic nematode genes. RNAi can be induced in M. incognita by feeding, with variable silencing efficiencies depending on the gene target35,36. M. incognita has many genes of the C. elegans RNAi pathway, including components of the amplification complex (ego-1, rrf-1, rrf-2 and rrf-3). However, we found no homologs of sid-1, sid-2, rsd-2 and rsd-6, which are genes involved in systemic RNAi and double-stranded RNA spreading to surrounding cells (Fig. 4, Supplementary Data, section 7.11; Supplementary Table 25 online). These genes are also absent from B. malayi7 and Haemonchus contortus37, suggesting that systematic RNAi may spread through the action of novel or poorly conserved factors. We retrieved 2,958 C. elegans genes having a lethal RNAi phenotype and searched for orthologs in M. incognita. Among the 1,083 OrthoMCL families identified, 148 (containing 344 M. incognita genes) appear to be nematode specific (Supplementary Data, section 7.12). Because of their lethal RNAi phenotype and distinctive sequence properties, these genes provide an attractive set of new antiparasite drug targets.

(a) Comparison of the RNAi pathway genes of C. elegans and M. incognita. A gray background indicates that at least one homologous gene was found in M. incognita, and a white background indicates that no homologous gene was found in M. incognita. (b) Distribution of orthologs to C. elegans lethal RNAi genes (Ce, black) between M. incognita (Mi, red), C. briggsae and B. malayi (Cb & Bm, green), D. melanogaster and three fungi, N. crassa, G. zea and M. grisea (Dm & 3 fungi, gray) using OrthoMCL. A yellow background indicates 148 nematode-only gene clusters.
Discussion
The genome of M. incognita has many traits that render it particularly attractive for studying the fundamentals of plant parasitism in the Nematoda. One remarkable feature is that most of the genome is composed of pairs of homologous segments that may denote former diverged alleles. This suggests that M. incognita is evolving without sex toward effective haploidy through the Meselson effect38,39,40. As the M. incognita genome is the first one sequenced and assembled for a strictly parthenogenetic species, we expect that its comparison with sexual nematode genomes will shed light on mechanisms leading to its peculiar structure. Functional divergence between ancient alleles of genes involved in the host-parasite interface could explain the extremely wide host range and geographic distribution of this polyphagous nematode. Analysis of the gene content of M. incognita revealed a suite of plant cell wall–degrading enzymes, which has no equivalent in any animal studied to date. The striking similarity of these enzymes to bacterial homologs suggests that these genes were acquired by multiple HGT events. Just as many instances of bacterial HGT involve sets of genes implicated in adaptations to new hosts or food sources, the candidate HGT events in M. incognita involve genes with potential roles in interactions with hosts. The alternative hypothesis—that these genes were acquired vertically from a common ancestor of bacteria and nematodes and lost in most eukaryote lineages—appears less parsimonious. Other singularities encompass M. incognita-restricted secreted proteins or lineage-specific expansions and/or reductions that may play roles in host-parasite interaction.
Transcriptional profiling, proteomic analysis and high throughput RNAi strategies are in progress and will lead to a deeper understanding of the processes by which a nematode causes plant disease. Combining such knowledge with functional genomic data from the model host plant A. thaliana should provide new insights into the intimate molecular dialog governing plant-nematode interactions and allow the further development of target-specific strategies to limit crop damage. Through the use of comparative genomics, the availability of free-living, animal- and plant-parasitic nematode genomes should provide new insights into parasitism and niche adaptation.
Methods
Strain and DNA extraction.
We used the M. incognita strain 'Morelos' from the root-knot nematode collection held at INRA (Institut National de la Recherche Agronomique) Sophia Antipolis, France. Nematode eggs were collected in a sterile manner from tomato roots and checked for the presence of plant material contaminants. DNA was extracted as described in Supplementary Methods, section 8.1 online.
Genome sequencing and assembly.
We obtained paired-end sequences from plasmid and BAC libraries with the Sanger dideoxynucleotide technology on ABI3730xl DNA analyzers. The 1,000,873 individual reads were assembled in 2,817 supercontigs using Arachne8 (Supplementary Methods, section 8.2; Supplementary Table 26 online).
Genome structure, operons and noncoding elements.
The assembled genome was searched for repetitive and non-coding elements. Scaffolds were aligned to determine pairs and triplets of allelic-like regions. Gene positions along scaffolds were used to predict clusters of genes forming putative operons (Supplementary Methods, section 8.3–8.7).
Prediction of protein coding genes.
Gene predictions were performed using EuGene14, optimized for M. incognita models and tested on a data set of 230 nonredundant, full-length cDNAs. Translation starts and splice sites were predicted by SpliceMachine41. Available M. incognita ESTs were aligned on the genome using GenomeThreader42. Similarities to C. elegans and other species' protein, genome and EST sequences were identified using BLAST43. Repetitive sequences were masked using RepeatMasker (http://repeatmasker.org/, Supplementary Methods, section 8.8; Supplementary Fig. 15 online).
Automatic functional annotation.
Protein domains were searched with InterproScan44. We also submitted proteins from seven additional species to the same InterproScan search. We included three other nematodes (C. elegans, C. briggsae and B. malayi), the fruitfly (D. melanogaster) and three fungi (Magnaporthe grisea, Gibberella zea and Neurospora crassa). To identify clusters of orthologous genes between M. incognita and the seven additional species, we used OrthoMCL15 (Supplementary Methods, section 8.9).
Expert functional annotation.
The collection of predicted protein coding genes was manually annotated by a consortium of laboratories. Each laboratory focused on a particular process or gene family relevant to the different aspects of M. incognita biology. Patterns of presence and/or absence and expansion and/or reduction in comparison to C. elegans, and other species were examined. The quality of predicted genes was manually checked and a functional annotation was proposed accordingly (Supplementary Methods, sections 8.10–8.20). A genome browser and additional information on the project are available from http://meloidogyne.toulouse.inra.fr/.
Accession codes.
The 9,538 contigs resulting from the Meloidogyne incognita genome assembly and annotation were deposited in the EMBL/Genbank/DDBJ databases under accession numbers CABB01000001–CABB01009538.
Note: Supplementary information is available on the Nature Biotechnology website.
Accession codes
References
Trudgill, D.L. & Blok, V.C. Apomictic, polyphagous root-knot nematodes: exceptionally successful and damaging biotrophic root pathogens. Annu. Rev. Phytopathol. 39, 53–77 (2001).
Blaxter, M.L. Nematoda: genes, genomes and the evolution of parasitism. Adv. Parasitol. 54, 101–195 (2003).
Caillaud, M.C. et al. MAP65–3 Microtubule-associated protein is essential for nematode-induced giant cell ontogenesis in Arabidopsis. Plant Cell 20, 423–437 (2008).
Caillaud, M.C. et al. Root-knot nematodes manipulate plant cell functions during a compatible interaction. J. Plant Physiol. 165, 104–113 (2008).
The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282, 2012–2018 (1998).
Stein, L.D. et al. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1, E45 (2003).
Ghedin, E. et al. Draft genome of the filarial nematode parasite Brugia malayi. Science 317, 1756–1760 (2007).
Jaffe, D.B. et al. Whole-genome sequence assembly for mammalian genomes: Arachne 2. Genome Res. 13, 91–96 (2003).
Leroy, S., Duperray, C. & Morand, S. Flow cytometry for parasite nematode genome size measurement. Mol. Biochem. Parasitol. 128, 91–93 (2003).
Triantaphyllou, A.C. in An Advance Treatise on Meloidogyne vol. 1 (eds. Sasser, J.N. & Carter, C.C.) 113–126, (North Carolina State University Graphics, Raleigh, USA, 1985).
Castagnone-Sereno, P. Genetic variability and adaptive evolution in parthenogenetic root-knot nematodes. Heredity 96, 282–289 (2006).
Mark Welch, D.B., Cummings, M.P., Hillis, D.M. & Meselson, M. Divergent gene copies in the asexual class Bdelloidea (Rotifera) separated before the bdelloid radiation or within bdelloid families. Proc. Natl. Acad. Sci. USA 101, 1622–1625 (2004).
Piotte, C., Castagnone-Sereno, P., Bongiovanni, M., Dalmasso, A. & Abad, P. Cloning and characterization of two satellite DNAs in the low-C-value genome of the nematode Meloidogyne spp. Gene 138, 175–180 (1994).
Foissac, S. & Schiex, T. Integrating alternative splicing detection into gene prediction. BMC Bioinformatics 6, 25 (2005).
Li, L., Stoeckert, C.J. Jr. & Roos, D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Davis, E.L., Hussey, R.S. & Baum, T.J. Getting to the roots of parasitism by nematodes. Trends Parasitol. 20, 134–141 (2004).
Wei, Y.D. et al. Molecular cloning, expression, and enzymatic activity of a novel endogenous cellulase from the mulberry longicorn beetle, Apriona germari. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 145, 220–229 (2006).
Qin, L. et al. Plant degradation: a nematode expansin acting on plants. Nature 427, 30 (2004).
Lambert, K.N., Allen, K.D. & Sussex, I.M. Cloning and characterization of an esophageal-gland-specific chorismate mutase from the phytoparasitic nematode Meloidogyne javanica. Mol. Plant Microbe Interact. 12, 328–336 (1999).
Hotson, A. & Mudgett, M.B. Cysteine proteases in phytopathogenic bacteria: identification of plant targets and activation of innate immunity. Curr. Opin. Plant Biol. 7, 384–390 (2004).
Tang, X., Xiao, Y. & Zhou, J.M. Regulation of the type III secretion system in phytopathogenic bacteria. Mol. Plant Microbe Interact. 19, 1159–1166 (2006).
Huang, G. et al. A profile of putative parasitism genes expressed in the esophageal gland cells of the root-knot nematode Meloidogyne incognita. Mol. Plant Microbe Interact. 16, 376–381 (2003).
Lindblom, T.H. & Dodd, A.K. Xenobiotic detoxification in the nematode Caenorhabditis elegans. J. Exp. Zoolog. A Comp. Exp. Biol. 305, 720–730 (2006).
Menzel, R., Bogaert, T. & Achazi, R. A systematic gene expression screen of Caenorhabditis elegans cytochrome P450 genes reveals CYP35 as strongly xenobiotic inducible. Arch. Biochem. Biophys. 395, 158–168 (2001).
Ewbank, J.J. Signaling in the immune response. WormBook doi/10.1895/wormbook.1.83.1, <http://www.wormbook.org/> (2006).
Alegado, R.A. & Tan, M.W. Resistance to antimicrobial peptides contributes to persistence of Salmonella typhimurium in the C. elegans intestine. Cell Microbiol. 10, 1259–1273 (2008).
Paschinger, K., Gutternigg, M., Rendic, D. & Wilson, I.B. The N-glycosylation pattern of Caenorhabditis elegans. Carbohydr. Res. 343, 2041–2049 (2007).
Bertrand, S. et al. Evolutionary genomics of nuclear receptors: from 25 ancestral genes to derived endocrine systems. Mol. Biol. Evol. 21, 1923–1937 (2004).
Robinson-Rechavi, M., Maina, C.V., Gissendanner, C.R., Laudet, V. & Sluder, A. Explosive lineage-specific expansion of the orphan nuclear receptor HNF4 in nematodes. J. Mol. Evol. 60, 577–586 (2005).
Plowman, G.D., Sudarsanam, S., Bingham, J., Whyte, D. & Hunter, T. The protein kinases of Caenorhabditis elegans: a model for signal transduction in multicellular organisms. Proc. Natl. Acad. Sci. USA 96, 13603–13610 (1999).
Robertson, H.M. & Thomas, J.H. The putative chemoreceptor families of C. elegans. WormBook doi/10.1895/wormbook.1.66.1, <http://www.wormbook.org/> (2006).
Marks, N.J. & Maule, A.G. in Neuropeptide Systems as Targets for Parasite and Pest Control (eds. Geary, T.G. & Maule, A.G.) (Landes Bioscience/Eurekah.com, Austin, TX, USA, 2008).
Zarkower, D. Somatic sex determination. WormBook doi/10.1895/wormbook.1.84.1, <http://www.wormbook.org/> (2006).
Papadopoulou, J. & Triantaphyllou, A.C. Sex-determinantion in Meloidogyne incognita and anatomical evidence of sexual reversal. J. Nematol. 14, 549–566 (1982).
Rosso, M.N., Dubrana, M.P., Cimbolini, N., Jaubert, S. & Abad, P. Application of RNA interference to root-knot nematode genes encoding esophageal gland proteins. Mol. Plant Microbe Interact. 18, 615–620 (2005).
Huang, G., Allen, R., Davis, E.L., Baum, T.J. & Hussey, R.S. Engineering broad root-knot resistance in transgenic plants by RNAi silencing of a conserved and essential root-knot nematode parasitism gene. Proc. Natl. Acad. Sci. USA 103, 14302–14306 (2006).
Zawadzki, J.L., Presidente, P.J., Meeusen, E.N. & De Veer, M.J. RNAi in Haemonchus contortus: a potential method for target validation. Trends Parasitol. 22, 495–499 (2006).
Birky, C.W. Jr. Bdelloid rotifers revisited. Proc. Natl. Acad. Sci. USA 101, 2651–2652 (2004).
Mark Welch, D. & Meselson, M. Evidence for the evolution of bdelloid rotifers without sexual reproduction or genetic exchange. Science 288, 1211–1215 (2000).
Mark Welch, D.B., Mark Welch, J.L. & Meselson, M. Evidence for degenerate tetraploidy in bdelloid rotifers. Proc. Natl. Acad. Sci. USA 105, 5145–5149 (2008).
Degroeve, S., Saeys, Y., De Baets, B., Rouze, P. & Van de Peer, Y. SpliceMachine: predicting splice sites from high-dimensional local context representations. Bioinformatics 21, 1332–1338 (2005).
Gremme, G., Brendel, V., Sparks, M.E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 47, 965–978 (2005).
Altschul, S.F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120 (2005).
Hillier, L.W. et al. Genomics in C. elegans: so many genes, such a little worm. Genome Res. 15, 1651–1660 (2005).
Acknowledgements
SCRI laboratory (V.C.B. and J.T.J.) received funding from the Scottish Government. This work benefited from links funded via COST Action 872. G.V.M. and V.L. are supported by ARC, CNRS, EMBO, MENRT and Region Rhone-Alpes. G.V.M., M.R.-R. and V.L. are also funded by the EU Cascade Network of Excellence and the integrated project Crescendo. M.-C.C. is supported by MENRT. We thank Philippe Lecomte for critical reading of the manuscript and all our collaborators from the “Plant-Nematode interaction” team of INRA Sophia Antipolis for technical help and support.
Author information
Authors and Affiliations
Contributions
P.A. and J.G. contributed equally as first authors. J.-M.A., P.C.-S., E.G.J.D., E.D. and L.P.-B. contributed equally as second authors. T.J.B., M.B., T.B-Z., E.L.D., J.J.E., B.F., E.G., B.H., J.T.J., V.L., A.G.M., H.Q., M.-N.R., T.S., G.S., J.W. and P.W. contributed equally as senior authors. P.A., M.B., P.C.-S. and E.G.J.D. wrote the manuscript with input from J.T.J. and A.G.M. For biological material, contributions were as follows. F.D., M.M. and L.P.-B. for strain growth, control and selection and DNA extraction. P.A., M.-C.C., F.D., E.D., B.F., M.-N.R. and L.P.-B. for cDNA libraries and EST data. For genome sequencing and assembly, contributions were as follows. B.S., E.U., J.P., V.A. for sequencing. C.J. for assembly. C.D. for cDNA clustering and library analyses. J.-M.A., O.J., C.J., F.A. for bioinformatics of allelism characterization. J.W. and P.W. supervision and coordination of the sequencing. For genome structure and organization, contributions were as follows. P.C.-S., T.F., H.Q. and D.S. for repetitive and transposable elements. J.G., E.S. for rRNAs, tRNAs, miRNAs. M.B. for operonic structures. M.-N.R., E.S. and C.V.G. for splice leaders (SL). For in-silico global genome analysis, contributions were as follows. E.D., J.G. and T.S. for gene predictions, automatic functional annotation, databases and bioinformatics. E.D. and B.F. for global protein set comparative analysis. Proteome expert annotation was as follows: P.M.C., E.G.J.D. and B.H., for Carbohydrate-Active enZymes. P.C.-S. and E.G. for proteases. M.-C.C., E.L.D., M.E., B.F., E.G.J.D., E.D., E.G., J.T.J., N.H., L.P.-B., G.S. and T.T. for candidate nematode parasitism and pioneer genes. P.A., T.B.-Z., E.G.J.D., E.D., J.J.E., J.V.G., G.P. and M.-N.R. for protection against plant defenses and immune system. V.L., G.V.M. and M.R.-R. for nuclear receptors. T.J.B., T.H. and T.R.M. for the kinome. E.G.J.D. and L.P.-B. for GPCRs. T.B.-Z., F.D.L., P.L. and P.V. for collagen. A.G.M. and P.M.V. for neuropeptides. J.T.J. for sex determination. V.C.B., E.G.J.D. and L.P.-B. for RNAi pathway and lethal RNAi phenotypes.
Corresponding author
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–15, Tables 1–26, Methods, Data (PDF 2367 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike license (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This license does not permit commercial exploitation, and derivative works must be licensed under the same or similar license.
About this article
Cite this article
Abad, P., Gouzy, J., Aury, JM. et al. Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat Biotechnol 26, 909–915 (2008). https://doi.org/10.1038/nbt.1482
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.1482
This article is cited by
-
In silico secretome analyses of the polyphagous root-knot nematode Meloidogyne javanica: a resource for studying M. javanica secreted proteins
BMC Genomics (2023)
-
Integrated management of Meloidogyne graminicola in nursery beds of rice
Indian Phytopathology (2023)
-
Developing Nano Silicon-Salicylic Acid Complex for Inducing Root-Knot Nematode Resistance in Tomato
Silicon (2023)
-
Control Phytophagous Nematodes By Engineering Phytosterol Dealkylation Caenorhabditis elegans as a Model
Molecular Biotechnology (2023)
-
Profile of Prof. Ke-Qin Zhang
Science China Life Sciences (2023)
