
Abstract
The shape, elongation, division and sporulation (SEDS) proteins are a large family of ubiquitous and essential transmembrane enzymes with critical roles in bacterial cell wall biology. The exact function of SEDS proteins was for a long time poorly understood, but recent work1,2,3 has revealed that the prototypical SEDS family member RodA is a peptidoglycan polymerase—a role previously attributed exclusively to members of the penicillin-binding protein family4. This discovery has made RodA and other SEDS proteins promising targets for the development of next-generation antibiotics. However, little is known regarding the molecular basis of SEDS activity, and no structural data are available for RodA or any homologue thereof. Here we report the crystal structure of Thermus thermophilus RodA at a resolution of 2.9 Å, determined using evolutionary covariance-based fold prediction to enable molecular replacement. The structure reveals a ten-pass transmembrane fold with large extracellular loops, one of which is partially disordered. The protein contains a highly conserved cavity in the transmembrane domain, reminiscent of ligand-binding sites in transmembrane receptors. Mutagenesis experiments in Bacillus subtilis and Escherichia coli show that perturbation of this cavity abolishes RodA function both in vitro and in vivo, indicating that this cavity is catalytically essential. These results provide a framework for understanding bacterial cell wall synthesis and SEDS protein function.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Meeske, A. J. et al. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537, 634–638 (2016)
Cho, H. et al. Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat. Microbiol. 1, 16172 (2016)
Emami, K. et al. RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat. Microbiol. 2, 16253 (2017)
Goffin, C. & Ghuysen, J.-M. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 62, 1079–1093 (1998)
McPherson, D. C. & Popham, D. L. Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilis. J. Bacteriol. 185, 1423–1431 (2003)
Packiam, M., Weinrick, B., Jacobs, W. R., Jr & Maurelli, A. T. Structural characterization of muropeptides from Chlamydia trachomatis peptidoglycan by mass spectrometry resolves ‘chlamydial anomaly’. Proc. Natl Acad. Sci. USA 112, 11660–11665 (2015)
Otten, C., Brilli, M., Vollmer, W., Viollier, P. H. & Salje, J. Peptidoglycan in obligate intracellular bacteria. Mol. Microbiol. 107, 142–163 (2018)
DiMaio, F. et al. Improved molecular replacement by density- and energy-guided protein structure optimization. Nature 473, 540–543 (2011)
Hopf, T. A. et al. Three-dimensional structures of membrane proteins from genomic sequencing. Cell 149, 1607–1621 (2012)
Hopf, T. A. et al. Sequence co-evolution gives 3D contacts and structures of protein complexes. eLife 3, e03430 (2014)
Marks, D. S. et al. Protein 3D structure computed from evolutionary sequence variation. PLoS ONE 6, e28766 (2011)
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007)
DiMaio, F. et al. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods 10, 1102–1104 (2013)
Qian, B. et al. High-resolution structure prediction and the crystallographic phase problem. Nature 450, 259–264 (2007)
Rigden, D. J., Keegan, R. M. & Winn, M. D. Molecular replacement using ab initio polyalanine models generated with ROSETTA. Acta Crystallogr. D 64, 1288–1291 (2008)
McCoy, A. J. et al. Ab initio solution of macromolecular crystal structures without direct methods. Proc. Natl Acad. Sci. USA 114, 3637–3641 (2017)
Strop, P., Brzustowicz, M. R. & Brunger, A. T. Ab initio molecular-replacement phasing for symmetric helical membrane proteins. Acta Crystallogr. D 63, 188–196 (2007)
Holm, L. & Rosenström, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010)
Mohammadi, T. et al. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 30, 1425–1432 (2011)
Hopf, T. A. et al. Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 35, 128–135 (2017)
Ovchinnikov, S. et al. Large-scale determination of previously unsolved protein structures using evolutionary information. eLife 4, e09248 (2015)
Leclercq, S. et al. Interplay between penicillin-binding proteins and SEDS proteins promotes bacterial cell wall synthesis. Sci. Rep. 7, 43306 (2017)
Caffrey, M. & Cherezov, V. Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 4, 706–731 (2009)
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010)
Johnson, L. S., Eddy, S. R. & Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431 (2010)
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B. & Wu, C. H. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932 (2015)
Ekeberg, M., Lövkvist, C., Lan, Y., Weigt, M. & Aurell, E. Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models. Phys. Rev. E 87, 012707 (2013)
Balakrishnan, S., Kamisetty, H., Carbonell, J. G., Lee, S.-I. & Langmead, C. J. Learning generative models for protein fold families. Proteins 79, 1061–1078 (2011)
Toth-Petroczy, A. et al. Structured states of disordered proteins from genomic sequences. Cell 167, 158–170.e12 (2016)
Jones, D. T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 (1999)
Käll, L., Krogh, A. & Sonnhammer, E. L. L. Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 35, W429–W432 (2007)
Brunger, A. T. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 (2007)
Brünger, A. T. et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 (1998)
Marks, D. S., Hopf, T. A. & Sander, C. Protein structure prediction from sequence variation. Nat. Biotechnol. 30, 1072–1080 (2012)
Bunkóczi, G. & Read, R. J. Improvement of molecular-replacement models with Sculptor. Acta Crystallogr. D 67, 303–312 (2011)
Schwarzenbacher, R., Godzik, A., Grzechnik, S. K. & Jaroszewski, L. The importance of alignment accuracy for molecular replacement. Acta Crystallogr. D 60, 1229–1236 (2004)
Schrodinger. The PyMOL Molecular Graphics System, Version 1.3r1 (Schrodinger, 2010)
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004)
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 68, 352–367 (2012)
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 66, 12–21 (2010)
Morin, A. et al. Collaboration gets the most out of software. eLife 2, e01456 (2013)
The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017)
Pakseresht, N. et al. Assembly information services in the European Nucleotide Archive. Nucleic Acids Res. 42, D38–D43 (2014)
Ashkenazy, H., Erez, E., Martz, E., Pupko, T. & Ben-Tal, N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 (2010)
Webb, B. & Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 54, 5.6.1–5.6.37 (2016)
Youngman, P. J., Perkins, J. B. & Losick, R. Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proc. Natl Acad. Sci. USA 80, 2305–2309 (1983)
Qiao, Y. et al. Lipid II overproduction allows direct assay of transpeptidase inhibition by β-lactams. Nat. Chem. Biol. 13, 793–798 (2017)
Qiao, Y. et al. Detection of lipid-linked peptidoglycan precursors by exploiting an unexpected transpeptidase reaction. J. Am. Chem. Soc. 136, 14678–14681 (2014)
Acknowledgements
Financial support for the work was provided by NIH grant U19AI109764 (A.C.K., D.Z.R., T.G.B., S.W. and D. K.), NIH grant R01GM106303 (D.S.M.) and a CIHR doctoral research award to P.D.A.R. We thank Advanced Photon Source GM/CA beamline staff for technical support during X-ray data collection, K. Arnett (Harvard Center for Macromolecular Interactions) for support of circular dichroism experiments and C. Sander for discussions.
Author information
Authors and Affiliations
Contributions
M.S. and A.J.M. performed expression screening experiments, and M.S. performed large-scale purification and crystallization of RodA as well as enzyme assays and circular dichroism spectroscopy. Additional input regarding enzyme assays was provided by P.D.A.R, V.S., D.K. and S.W. The structure was solved and refined by M.S. and A.C.K. using evolutionary coupling-derived models developed by K.B., A.G.G., T.A.H. and D.S.M. Assessment of RodA mutant phenotypes was conducted by G.D. and P.D.A.R. with supervision from T.G.B. and D.Z.R. Overall project supervision was performed by A.C.K. with input from T.G.B. and D.Z.R. The manuscript was written by M.S. and A.C.K. with input from other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks R. Read, K. Young and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Representative electron density.
a–c, Simulated annealing composite omit 2Fo − Fc electron density map of T. thermophilus RodA contoured at 1.0σ within a 2.0 Å radius of atoms shown. d, The same map contoured at 1.0σ and coloured blue and green for RodA and water molecules, respectively. The modelled water molecules are shown as red spheres. e, The same map contoured at 1.0σ within a 3.0 Å radius RodA (shown in blue) and the same map contoured at 1.0σ for monoolein (shown in black).
Extended Data Figure 2 Comparison of RodA wild type and RodA(D255A) structures.
Structures of wild-type (green) and D255A (blue) RodA are shown viewed parallel to the membrane (left) and from the extracytoplasmic side (right). The Cα atom of residue 255 for each structure is shown as a sphere, and coloured pink (wild type) or yellow (D255A). The dashed lines represent the disordered residues 189–227 and 237–251 in both structures. The two structures are essentially identical, with a Cα r.m.s.d. of 0.1 Å.
Extended Data Figure 3 RodA sequence conservation.
The results of an alignment of 506 RodA sequences from diverse bacterial taxa, with representative examples displayed. Residues with 98%, 80% and 60% similarity across all 506 sequences are shown in black, grey and light grey, respectively. Secondary structure elements are shown above the alignment on the basis of the T. thermophilus RodA crystal structure and JPRED analysis of the portions of ECL4 that were not modelled in the structure.
Extended Data Figure 4 RodA evolutionary couplings.
a, The refined crystal structure of T. thermophilus RodA (shown in light blue) is in close agreement with its evolutionary couplings. Green, yellow and red lines between residues represent regions of the structure that are less than 5 Å, between 5 and 10 Å, and greater than 10 Å of the predicted evolutionary couplings, respectively. b, The partially disordered ECL4 in the refined structure is strongly coupled evolutionarily to the transmembrane domain of RodA. A representation of predicted intra- and inter-domain evolutionary couplings for ECL4 is mapped onto a EVfold model (shown in grey).
Extended Data Figure 5 Sequence conservation of T. thermophilus RodA.
a, Surface and ribbon representation of RodA (top and bottom, respectively). Analysis was performed using Consurf, coloured in a scale from teal (poorly conserved) to magenta (highly conserved). A bound lipid modelled as monoolein is shown in yellow sticks. The dashed lines represent disordered residues 189–227 and 237–251 in ECL4. b, The bound lipid (shown as spheres) is surrounded by many aliphatic amino acids (shown as sticks). c, Extracytoplasmic view of the water-filled central cavity and its proximity to the bound lipid molecule.
Extended Data Figure 6 Circular dichroism spectroscopy analysis of purified B. subtilis RodA variants.
Circular dichroism measurements of wild-type RodA as well as RodA(D280A), RodA(E117K/K120E) and RodA(I262S) indicate that the overall folds of all four forms are comparable and display characteristic α-helical peaks at 208 and 222 nm. For each panel, the circular dichroic spectra of wild-type and the indicated mutant RodA are shown in black and blue, respectively. The corresponding T. thermophilus numbering for each mutant is shown in parentheses. Experiments were repeated independently twice with similar results.
Extended Data Figure 7 Mutagenic analysis of B. subtilis RodA function in vivo.
Micrographs of B. subtilis strains harbouring an IPTG-inducible allele of wild-type rodA, and wild-type or mutant PxylA-rodA. Expression was induced by growing cells in the presence of 10 μM IPTG and 10 mM xylose. The bacterial cytosol is indicated by intracellular mCherry expression (Ppen-mCherry). Mutants in the central cavity show particularly deleterious phenotypes in this dominant negative assay. Experiments were repeated independently 2–3 times with similar results. A mutation made in the predicted RodA–bPBP interface does not prevent peptidoglycan polymerization in vitro (lower left panel), representative of two independent experiments.
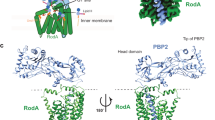
Extended Data Figure 8 Evolutionary coupling analysis for the RodA–PBP2 complex.
Evolutionary co-variation maps highlighting 693 couplings for PBP2 (top left panel), and 362 couplings for RodA (bottom right panel) using a 95% confidence threshold. These maps display good correlation to the crystal structure of RodA and homology models of PBP2. The top right and lower bottom panels depict the 19 predicted inter-protein contacts between RodA and PBP2 using the same 95% confidence threshold.
Supplementary information
Supplementary Figure 1
This file contains the uncropped gels. (PDF 859 kb)
Rights and permissions
About this article

Cite this article
Sjodt, M., Brock, K., Dobihal, G. et al. Structure of the peptidoglycan polymerase RodA resolved by evolutionary coupling analysis. Nature 556, 118–121 (2018). https://doi.org/10.1038/nature25985
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25985
This article is cited by
-
Structural insights into the activation of the divisome complex FtsWIQLB
Cell Discovery (2024)
-
Diversity of sugar-diphospholipid-utilizing glycosyltransferase families
Communications Biology (2024)
-
Allosteric activation of cell wall synthesis during bacterial growth
Nature Communications (2023)
-
The relaxin receptor RXFP1 signals through a mechanism of autoinhibition
Nature Chemical Biology (2023)
-
Cryo-EM structure of the bacterial divisome core complex and antibiotic target FtsWIQBL
Nature Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
