
Abstract
Distant-acting tissue-specific enhancers, which regulate gene expression, vastly outnumber protein-coding genes in mammalian genomes, but the functional importance of this regulatory complexity remains unclear1,2. Here we show that the pervasive presence of multiple enhancers with similar activities near the same gene confers phenotypic robustness to loss-of-function mutations in individual enhancers. We used genome editing to create 23 mouse deletion lines and inter-crosses, including both single and combinatorial enhancer deletions at seven distinct loci required for limb development. Unexpectedly, none of the ten deletions of individual enhancers caused noticeable changes in limb morphology. By contrast, the removal of pairs of limb enhancers near the same gene resulted in discernible phenotypes, indicating that enhancers function redundantly in establishing normal morphology. In a genetic background sensitized by reduced baseline expression of the target gene, even single enhancer deletions caused limb abnormalities, suggesting that functional redundancy is conferred by additive effects of enhancers on gene expression levels. A genome-wide analysis integrating epigenomic and transcriptomic data from 29 developmental mouse tissues revealed that mammalian genes are very commonly associated with multiple enhancers that have similar spatiotemporal activity. Systematic exploration of three representative developmental structures (limb, brain and heart) uncovered more than one thousand cases in which five or more enhancers with redundant activity patterns were found near the same gene. Together, our data indicate that enhancer redundancy is a remarkably widespread feature of mammalian genomes that provides an effective regulatory buffer to prevent deleterious phenotypic consequences upon the loss of individual enhancers.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Accession codes
References
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012)
Long, H. K., Prescott, S. L. & Wysocka, J. Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170–1187 (2016)
Andrey, G. & Mundlos, S. The three-dimensional genome: regulating gene expression during pluripotency and development. Development 144, 3646–3658 (2017)
Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M. & Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development 132, 797–803 (2005)
Menke, D. B., Guenther, C. & Kingsley, D. M. Dual hindlimb control elements in the Tbx4 gene and region-specific control of bone size in vertebrate limbs. Development 135, 2543–2553 (2008)
Shim, S., Kwan, K. Y., Li, M., Lefebvre, V. & Sestan, N. Cis-regulatory control of corticospinal system development and evolution. Nature 486, 74–79 (2012)
Hay, D. et al. Genetic dissection of the α-globin super-enhancer in vivo. Nat. Genet. 48, 895–903 (2016)
Frankel, N. et al. Phenotypic robustness conferred by apparently redundant transcriptional enhancers. Nature 466, 490–493 (2010)
Perry, M. W., Boettiger, A. N., Bothma, J. P. & Levine, M. Shadow enhancers foster robustness of Drosophila gastrulation. Curr. Biol. 20, 1562–1567 (2010)
Montavon, T. et al. A regulatory archipelago controls Hox genes transcription in digits. Cell 147, 1132–1145 (2011)
Petit, F., Sears, K. E. & Ahituv, N. Limb development: a paradigm of gene regulation. Nat. Rev. Genet. 18, 245–258 (2017)
Zeller, R., López-Ríos, J. & Zuniga, A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat. Rev. Genet. 10, 845–858 (2009)
Visel, A., Minovitsky, S., Dubchak, I. & Pennacchio, L. A. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007)
Pennacchio, L. A. et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006)
Attanasio, C. et al. Fine tuning of craniofacial morphology by distant-acting enhancers. Science 342, 1241006 (2013)
Osterwalder, M. et al. HAND2 targets define a network of transcriptional regulators that compartmentalize the early limb bud mesenchyme. Dev. Cell 31, 345–357 (2014)
Rosin, J. M., Abassah-Oppong, S. & Cobb, J. Comparative transgenic analysis of enhancers from the human SHOX and mouse Shox2 genomic regions. Hum. Mol. Genet. 22, 3063–3076 (2013)
Cobb, J., Dierich, A., Huss-Garcia, Y. & Duboule, D. A mouse model for human short-stature syndromes identifies Shox2 as an upstream regulator of Runx2 during long-bone development. Proc. Natl Acad. Sci. USA 103, 4511–4515 (2006)
Akiyama, H., Chaboissier, M. C., Martin, J. F., Schedl, A. & de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828 (2002)
Kawakami, Y. et al. Sall genes regulate region-specific morphogenesis in the mouse limb by modulating Hox activities. Development 136, 585–594 (2009)
Min, H. et al. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 12, 3156–3161 (1998)
Andrey, G. et al. Characterization of hundreds of regulatory landscapes in developing limbs reveals two regimes of chromatin folding. Genome Res. 27, 223–233 (2017)
Hui, C. C. & Joyner, A. L. A mouse model of Greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 3, 241–246 (1993)
Lopez-Rios, J. et al. GLI3 constrains digit number by controlling both progenitor proliferation and BMP-dependent exit to chondrogenesis. Dev. Cell 22, 837–848 (2012)
Ye, W. et al. A unique stylopod patterning mechanism by Shox2-controlled osteogenesis. Development 143, 2548–2560 (2016)
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012)
Marinic´, M., Aktas, T., Ruf, S. & Spitz, F. An integrated holo-enhancer unit defines tissue and gene specificity of the Fgf8 regulatory landscape. Dev. Cell 24, 530–542 (2013)
Visel, A. et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895–908 (2013)
Lam, D. D. et al. Partially redundant enhancers cooperatively maintain mammalian pomc expression above a critical functional threshold. PLoS Genet. 11, e1004935 (2015)
Yao, Y. et al. Cis-regulatory architecture of a brain signaling center predates the origin of chordates. Nat. Genet. 48, 575–580 (2016)
Antosova, B. et al. The gene regulatory network of lens induction is wired through Meis-dependent shadow enhancers of Pax6. PLoS Genet. 12, e1006441 (2016)
Will, A. J. et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat. Genet. 49, 1539–1545 (2017)
Hong, J. W., Hendrix, D. A. & Levine, M. S. Shadow enhancers as a source of evolutionary novelty. Science 321, 1314 (2008)
Barolo, S. Shadow enhancers: frequently asked questions about distributed cis-regulatory information and enhancer redundancy. BioEssays 34, 135–141 (2012)
Cannavò, E. et al. Shadow enhancers are pervasive features of developmental regulatory networks. Curr. Biol. 26, 38–51 (2016)
Yanagisawa, H., Clouthier, D. E., Richardson, J. A., Charité, J. & Olson, E. N. Targeted deletion of a branchial arch-specific enhancer reveals a role of dHAND in craniofacial development. Development 130, 1069–1078 (2003)
Fulco, C. P. et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773 (2016)
Lettice, L. A., Hill, A. E., Devenney, P. S. & Hill, R. E. Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet. 17, 978–985 (2008)
Lupiáñez, D. G. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015)
Kothary, R. et al. Inducible expression of an hsp68-lacZ hybrid gene in transgenic mice. Development 105, 707–714 (1989)
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009)
Mo, A. et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384 (2015)
Yang, H. et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379 (2013)
Yang, H., Wang, H. & Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protocols 9, 1956–1968 (2014)
Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M. & Valen, E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401–W407 (2014)
Kvon, E. Z. et al. Progressive loss of function in a limb enhancer during snake evolution. Cell 167, 633–642 (2016)
Ovchinnikov, D. Alcian blue/alizarin red staining of cartilage and bone in mouse. Cold Spring Harb. Protoc. 2009, prot5170 (2009)
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nat. Protocols 3, 1101–1108 (2008)
Dickel, D. E. et al. Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun. 7, 12923 (2016)
Nord, A. S. et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531 (2013)
Trapnell, C. et al. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010)
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015)
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010)
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009)
Zhang, Y. et al. Model-based analysis of ChIP–seq (MACS). Genome Biol. 9, R137 (2008)
Jalili, V., Matteucci, M., Masseroli, M. & Morelli, M. J. Using combined evidence from replicates to evaluate ChIP–seq peaks. Bioinformatics 31, 2761–2769 (2015)
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013)
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012)
Yanai, I. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659 (2005)
Bult, C. J., Eppig, J. T., Blake, J. A., Kadin, J. A. & Richardson, J. E. Mouse genome database 2016. Nucleic Acids Res. 44, D840–D847 (2016)
Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056 (2015)
Speir, M. L. et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 44, D717–D725 (2016)
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010)
Gordon, C. T. et al. Identification of novel craniofacial regulatory domains located far upstream of SOX9 and disrupted in Pierre Robin sequence. Hum. Mutat. 35, 1011–1020 (2014)
Li, Q. et al. A Gli silencer is required for robust repression of gremlin in the vertebrate limb bud. Development 141, 1906–1914 (2014)
Acknowledgements
This work was supported by National Institutes of Health grants R01HG003988, U54HG006997, R24HL123879 and UM1HL098166 (to A.V. and L.A.P.) and the University of Basel and the Novartis Foundation for Biomedical Research (to J.L.-R.). M.O. was supported by a Swiss National Science Foundation (SNSF) fellowship. We thank B. Ren for providing access to the ChIP–seq and RNA-seq data from ENCODE; J. Doudna for providing a plasmid containing a human-optimized Cas9 gene; W. Ye and Y. Chen for sharing the image of a Shox2-deficient limb skeleton (Fig. 3b); and the members of the L.A.P., A.V. and D.E.D. groups for technical advice and comments on the manuscript, in particular C. Spurrell and E. Kvon. Research was conducted at the E. O. Lawrence Berkeley National Laboratory and performed under Department of Energy Contract DE-AC02-05CH11231, University of California.
Author information
Authors and Affiliations
Contributions
M.O., D.E.D., A.V., and L.A.P. conceived the study. M.O., D.E.D., B.J.M., S.Y.A., E.A.L., Y.Z., I.P.-F., C.S.P., M.K., T.H.G., Q.T.P., A.N.H., J.A.A., and V.A. performed the genome editing and mouse phenotyping studies. I.B. and M.O. devised the computational framework, and I.B. performed the correlative analysis. V.T. performed in situ hybridization under the supervision of J.L.-R. Y.F.-Y. conducted the ChIP–seq and RNA-seq data analysis. M.O., D.E.D., A.V., and L.A.P. wrote the manuscript with input from the remaining authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 CRISPR deletion of ten limb enhancers and regulatory interaction landscape of associated target genes.
a–j, Left, representative activity patterns of the selected enhancers in mouse embryos at E11.5 (VISTA enhancer browser)13 and the respective genomic enhancer regions tested in transgenic assays (Tg, blue bar), along with the regions deleted in enhancer knockout mice (Del, red bar). Corresponding H3K27 acetylation patterns (green) in wild-type mouse embryonic forelimbs at E11.5 (this study) are depicted with open chromatin (ENCODE DHS in forelimbs at E11.5, purple) and the Placental Mammal basewise conservation track by PhyloP (Cons, blue/red). Scale bars, 500 bp. VISTA enhancer IDs (mm and hs numbers) are indicated on the left, with the distance of the enhancer from the transcriptional start site of the predicted target gene in the mouse genome. Numbers at the bottom right of each embryo indicate the reproducibility of the enhancer reporter assay. Arrowheads mark additional activity domains (other than limb): hs1262 (hindbrain, reproducibility: 5/6, also shown previously17), mm917 (dorsal root ganglion, 7/7) and hs1603 (nose, 7/7; and branchial arch, 5/7). Asterisk indicates potential craniofacial enhancer activity for mm636, which was observed in 3 of 9 embryos64. Right, PCR validation strategy and results for enhancer knockout lines. Red scissors indicate CRISPR-mediated deletion breakpoints. PCR was used to detect the wild-type (+) and enhancer deletion (Δ) alleles. Below, Sanger sequencing traces show the deletion breakpoints (indicated by the dashed line) for the enhancer knockout alleles. PCR genotyping results are shown with amplicon sizes indicated on the left (enhancer deletion allele in red). Primers (Ctrl or Ctrl2) amplifying an unrelated genomic region were included as a PCR positive control. See Supplementary Table 3 for all primer sequences and related PCR product sizes. k, Top, Hi-C interaction heat maps of topologically associated chromatin domains (mouse embryonic stem cell TADs)26. Bottom, selected enhancers (blue triangles) and their predicted target genes (TSS indicated as black bar). The Capture-C UCSC browser track (purple) illustrates three-dimensional chromatin interaction profiles from E11.5 embryonic limbs (3-kb window) using promoters of the predicted enhancer target genes as anchor points22. H3K27ac enrichment (green) in wild-type forelimbs at E11.5 (this study) is shown below. Six of the ten enhancers selected for deletion analysis display local Capture-C enrichment (*), indicating physical interaction with the predicted target gene promoter at E10.5 or E11.5, based on the stringent statistical approach (95th percentile threshold) applied in the original study22. Other genes present in the TAD are shown in grey.
Extended Data Figure 2 No major differences in expression of predicted target genes in individual enhancer knockouts.
a, Spatial enhancer activity domains (LacZ, see also Fig. 1b) are compared to mRNA expression domains (by in situ hybridization) of the predicted target genes in embryonic forelimbs and hindlimbs at E11.5. No significant changes in expression patterns were observed in enhancer knockouts compared to wild-type limbs, except in limbs lacking hs741, where a small subdomain of target gene expression was lost (red arrowhead marks loss of the posterior Shox2 domain in the distal limb, compared with black arrowhead in wild type). Transcript distribution was reproduced in at least n = 3 independent biological replicates. b, Quantitative real-time PCR using limbs of homozygous null (KO, red dots) and wild-type (Wt, blue dots) embryos at E11.5 reveals lack of significantly downregulated transcript levels of predicted enhancer target genes in nine out of ten cases. Box plots indicate median, interquartile values, range and individual biological replicates. Outliers are shown as circled data points. **P = 0.0012, unpaired, two-tailed t-test. n.s., not significant. Scale bars, 100 μm.
Extended Data Figure 3 Absence of obvious morphological abnormalities in limb enhancer knockouts.
Side-by-side comparison of enhancer knockout limb skeletons and wild-type littermate controls at E18.5. Neither forelimbs (this figure) nor hindlimbs (data not shown) of the enhancer knockout lines revealed any obvious morphological differences in comparison to wild-type littermates. Cartilage is stained blue and bone dark red. The number of embryos with normal limb phenotypes over the total number of homozygous-null embryos examined is shown in the bottom left. n represents number of independent biological replicates with similar results. Scale bar, 1 mm.
Extended Data Figure 4 Absence of compensatory enhancer signatures in limbs of enhancer knockout embryos.
a, Layered ChIP–seq H3K27 acetylation (ac) profiles surrounding the deleted enhancers and from wild-type (blue, n = 4 independent biological replicates) and enhancer knockout embryos (orange, at least n = 2 biological replicates). For all samples, E11.5 forelimb was profiled. For display, replicates were merged using bigWigMerge (UCSC tools) and normalized. Red triangles indicate the positions of individual enhancer deletions. b, H3K27ac enrichments in targeted regions marked by red triangles in a, showing the absence of H3K27ac at the deletion site in individual enhancer knockout (orange) compared to wild-type (blue) samples. Blue bars indicate locations of enhancer sequences. Dashed red lines demarcate the regions deleted by CRISPR. Vertebrate basewise conservation track by PhyloP (Cons) is shown.
Extended Data Figure 5 Transcriptional and phenotypic impact of dual enhancer deletions engineered by iterative CRISPR–Cas9 genome editing.
a–c, Top, enhancer pairs with overlapping limb activities (LacZ), coinciding with domains of predicted target gene expression visualized by in situ hybridization (ISH). For Sox9 enhancers, black arrowheads indicate overlapping domains. Schematics, double enhancer deletion strategy to delete the three enhancer pairs with overlapping activity (see Methods). Grey numbers indicate enhancer distance (kb) from the TSS. Bottom, Sanger sequencing verification of the secondary enhancer deletion. Deletion breakpoint is marked by the dashed line. Grey horizontal bars indicate bases present in the primary deletions (single enhancer knockout lines, see Extended Data Fig. 1a–j). Shox2- and Sox9-associated LacZ panels are also used in Extended Data Fig. 2. d, Gli3 transcript distribution in situ hybridization in wild-type (Wt) and mm1179/hs1586 DKO embryos. Arrowhead points to reduced Gli3 transcript in the anterior limb mesenchyme. Dashed line indicates dissected hand plate for RNA-seq. e, RNA-seq confirmed significantly reduced Gli3 expression in hand plates of DKO embryos but not individual enhancer knockout embryos (compared to wild-type hand plates). f, Unaffected hindlimb morphology in mm1179/hs1586 DKO embryos. Red arrowhead points to digit 1 duplication in forelimbs (see also Fig. 2). g, Shox2 expression (in situ hybridization) in forelimbs and hindlimbs of hs741/hs1262 DKO embryos. The distal-posterior domain (arrowhead) is dependent on hs741 (Extended Data Fig. 2a). h, Reduced Shox2 expression in forelimbs and hindlimbs of hs741/hs1262 DKO embryos (qPCR). Expression of the nearby Rsrc1 gene was unchanged. i, Left, representative limb skeletons of wild-type and hs741/hs1262 DKO embryos. Hu, humerus; Ul, ulna; Fe, femur; Ti, tibia. Right, mild but significant reduction in humerus ossification length (double arrows) in hs741/hs1262 DKO limb skeletons. ***P = 1.66 × 10−7 (two-tailed, unpaired t-test). j, Absence of evident differences in Sox9 expression or skeletal abnormalities in embryos lacking both the hs1467 and mm636 enhancers near Sox9. For in situ hybridization, transcript distribution was reproduced in at least n = 3 independent biological replicates. n represents number of independent biological replicates with similar results. For bar graphs and boxplots, individual biological replicates are shown as data points. Bar graphs illustrate mean and s.d. Box plot indicates median, interquartile values and range. ***P < 0.001; **P < 0.01 (two-tailed, unpaired t-test). n.s., not significant. Scale bars, 100 μm (white) and 500 μm (black).
Extended Data Figure 6 Cellular resolution of redundant Gli3 enhancer activities at the onset of digit formation.
a, b, Individual Gli3 enhancer activities as detected by immunofluorescence (mm1179, green; hs1586, red) in forelimbs of transgenic reporter embryos. Sox9 (grey) marks chondrogenic progenitors of the mesenchymal condensations forming digit primordia (digits 1–5, from anterior to posterior). c, d, Co-localization of mm1179 and hs1586 enhancer activities in hand plates of double enhancer transgenic embryos. Close-ups (right) show that the anterior mesenchyme (Fig. 2c) harbours many cells with dual enhancer activities (yellow). A fraction of double enhancer-positive cells carries the signature of Sox9 digit progenitors (white, bottom). n = 3 independent embryos per genotype were analysed, with similar results. Nuclei, detected via Hoechst staining, are blue. Scale bars, 100 μm (a, b); 50 μm (c, d).
Extended Data Figure 7 Generation of Gli3 and Shox2 knockout alleles and characterization of enhancer deletions in a sensitized background.
a, d, Top, schematic showing CRISPR–Cas9-mediated deletions used to generate Gli3 and Shox2 loss-of-function alleles. Genotyping primers used to validate targeted deletion events are indicated. Bottom, Sanger sequencing confirmation of deletion event, with grey and red dashed lines indicating breakpoints. Right, PCR genotyping examples with the size of the product specific for the deletion allele depicted in red (primers listed in Supplementary Table 3). b, In situ hybridization showing the gradual decrease in anterior Gli3 transcript in forelimbs of wild-type, Gli3Δ/+ and sensitized mm1179/hs1586 DKO (DKO/Gli3Δ) embryos. c, qPCR validation of Gli3 mRNA levels in forelimb hand plates from the genotypes shown in b. e, Shox2 expression (in situ hybridization) in forelimbs and hindlimbs of wild-type, Shox2Δ/+ and sensitized hs741/hs1262 DKO (DKO/Shox2Δ) embryos. Arrowheads point to the domains where Shox2 expression is nearly abolished in enhancer DKO/Shox2Δ embryos. f, qPCR revealing significantly downregulated Shox2 mRNA levels in hindlimbs of DKO/Shox2Δ compared to Shox2Δ/+ embryos. n indicates the number of independent biological replicates with similar results. Bar plots illustrate mean and s.d., with individual biological replicates shown. ***P < 0.001; *P < 0.05 (two-tailed, unpaired t-test). n.s., not significant. For in situ hybridization, transcript distribution was reproduced in at least n = 3 independent biological replicates. Scale bars, 100 μm.
Extended Data Figure 8 Limb phenotypes of individual and combinatorial Gli3 and Shox2 enhancer knockouts in the presence of reduced target gene dosage.
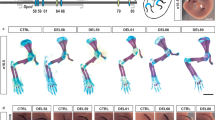
a, Skeletal phenotypes resulting from mm1179 and hs1586 enhancer deletions in combination with reduction to one copy of the Gli3 gene at E18.5. Genotypes are shown on the left with red crosses indicating elements deleted by CRISPR–Cas9. While forelimbs of Gli3Δ/+ embryos displayed bifurcated digit 1 terminal phalanges65, hindlimbs showed an extra toe structure but without detectable cartilage template. Four out of seven mm1179Δ/Gli3Δ embryos displayed additional bifurcation of digit 2 of the right forelimb (a), which suggests that removal of mm1179 reduces Gli3 levels in the anterior forelimb more than deletion of hs1586. An almost complete anterior extra toe formed in hindlimbs of embryos with single or dual enhancer deletions in the sensitized background (black asterisks). Loss of both Gli3 copies resulted in anterior hindlimb polydactyly with altered digit identities (red asterisks)24. b, Allelic series depicting shortening of the stylopod (humerus and femur) in limb skeletons with individual or combined hs741 and hs1262 enhancer deletions in a Shox2 sensitized condition (see also Fig. 3b). Stylopod ossification length (double arrows) appears less reduced in forelimbs (humerus, Hu) than in hindlimbs (femur, Fe) of embryos lacking the activity of both enhancers (hs741Δ, hs1262Δ/Shox2Δ). Tibia (Ti) and ulna (Ul) were normal in all genotypes examined. c, Humerus ossification length (normalized to ulna ossification length) is significantly reduced in embryos lacking either hs741 or hs1262 in the presence of only one copy of Shox2. In embryos lacking both enhancers in the sensitized background, significant shortening of humerus ossification is observed (compared to all other genotypes). n indicates the number of independent biological replicates with similar results. Box plots indicate median, interquartile values, range and individual biological replicates. ***P < 0.001; *P < 0.05 (two-tailed, unpaired t-test). Scale bars, 500 μm.
Extended Data Figure 9 A correlative framework to define enhancer–promoter associations across the mouse genome.
a, The TAD including the transcriptional regulators Tbx3, Tbx5 and Lhx5 illustrates the statistical framework to define enhancer–promoter associations genome-wide. For each predicted enhancer, correlation between its H3K27ac signal (blue arrowhead, blue-shaded heat map) with the mRNA expression profiles of every gene in the TAD (red-shaded heat map) across all available tissues and developmental stages was assessed. The enhancer was then assigned to the most highly correlated gene, Tbx3 in the case of enhancer 3. b, Schematic depicting the underlying statistical framework used to determine genome-wide enhancer–promoter interactions (see Methods). c, Activity pattern for the enhancers assigned to Tbx3, Tbx5 and Lhx5. Genomic coordinates are listed on the right. For each predicted enhancer–gene pair, Spearman’s correlation coefficient (SCC, n = 29) and the corresponding empirically estimated P value (from 1,000 random enhancer–gene pairings) are shown in Supplementary Table 11. d, Identifying genes with biased expression in embryonic limb, forebrain, or heart. Expression variability across 29 RNA-seq datasets from multiple tissues and developmental time points, measures of tissue specificity (Tau (τ), x-axis) and specific tissue-biased expression at E11.5 (y-axis) for each protein-coding gene were calculated (see Methods). Housekeeping genes were defined as displaying τ ≤ 0.4 and relative expression in the limb between the 5th and 95th percentiles. Tissue-biased genes were defined as showing τ ≥ 0.7 and relative expression higher than the 95th percentile. d, Distribution of enhancer numbers assigned to each gene, for the different gene categories. Genes with tissue-biased expression profiles were associated with a significantly higher number of enhancers than housekeeping genes. P = 4 × 10−121 (n = 553), P = 7 × 10−97 (n = 626) and P = 6 × 10−83 (n = 826) for limb, forebrain and heart biased genes, respectively (two-sided Mann–Whitney tests). n = 1,287 for housekeeping genes. Box plots indicate median, interquartile values and range. Outliers are shown as individual points.
Extended Data Figure 10 Enhancer redundancy as a widespread feature of developmental genes and robustness to the choice of thresholds used in the correlative approach.
a, b, Top, number of enhancers assigned to each gene through the correlative framework, with developmental transcription factors (TFs) showing biased expression in forebrain (a, blue dots) or heart (b, orange dots) indicated. Classification of tissue-biased developmental transcription factors is described in Methods. Genes with at least one assigned enhancer are displayed and sorted according to the number of assigned enhancers (left to right). Bottom, bar plot showing the total number of enhancers assigned to each of the transcription factors highlighted in the top panels. For each gene, a colour code shows the number of predicted enhancers assigned to that gene in the relevant tissue (a, heart; b, forebrain) at E11.5 (dark colour), in the relevant tissue at any other developmental stage included in the analysis (light colour), or in any other tissue (white). c, Estimated FDR (based on genome-wide permutations, see Methods) of observing a gene with five or more enhancers assigned to it, for increasingly larger correlation coefficients (0.25 to 0.75). The red solid line indicates an FDR of 0.05. The red arrow and the black dashed line highlight the lowest correlation coefficient (0.47, considering a step of 0.01) with an FDR ≤ 0.05 (FDR = 0.0495). d, Number of genes showing five or more enhancers assigned to them, for increasingly larger correlation coefficients (0.25 to 0.75). The total number of genes (SCC ≥ 0.25) along with the number of genes identified using the threshold set in c (SCC > = 0.47) is indicated (1,276 and 1,058, respectively; see Supplementary Tables 11, 12). e, Bubble plot showing the number of genes with five or more enhancers assigned to them, at increasingly higher correlation between enhancer and target gene expression (x-axis) and between enhancers assigned to the same gene (y-axis). f, Bubble plot displaying the fold-enrichment (linear) for developmental transcription factor genes among each set in c.
Supplementary information
Supplementary Tables
This file contains Supplementary Tables 1-4, 8, 10 and additional references. The file also contains full legends for Supplementary Tables 5, 6, 7, 9, 11 and 12. The Supplementary Tables are provided as separate Excel files in a zipped file. (PDF 373 kb)
Supplementary Tables
This file contains Supplementary Tables 5, 6, 7, 9, 11 and 12. The full Supplementary Table legends are provided separately in a PDF file. (ZIP 3409 kb)
Rights and permissions
About this article

Cite this article
Osterwalder, M., Barozzi, I., Tissières, V. et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243 (2018). https://doi.org/10.1038/nature25461
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25461
This article is cited by
-
Boundary stacking interactions enable cross-TAD enhancer–promoter communication during limb development
Nature Genetics (2024)
-
Omics-based construction of regulatory variants can be applied to help decipher pig liver-related traits
Communications Biology (2024)
-
Increased enhancer–promoter interactions during developmental enhancer activation in mammals
Nature Genetics (2024)
-
3D Enhancer–promoter networks provide predictive features for gene expression and coregulation in early embryonic lineages
Nature Structural & Molecular Biology (2024)
-
A distant global control region is essential for normal expression of anterior HOXA genes during mouse and human craniofacial development
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
