
Abstract
Salamanders serve as important tetrapod models for developmental, regeneration and evolutionary studies. An extensive molecular toolkit makes the Mexican axolotl (Ambystoma mexicanum) a key representative salamander for molecular investigations. Here we report the sequencing and assembly of the 32-gigabase-pair axolotl genome using an approach that combined long-read sequencing, optical mapping and development of a new genome assembler (MARVEL). We observed a size expansion of introns and intergenic regions, largely attributable to multiplication of long terminal repeat retroelements. We provide evidence that intron size in developmental genes is under constraint and that species-restricted genes may contribute to limb regeneration. The axolotl genome assembly does not contain the essential developmental gene Pax3. However, mutation of the axolotl Pax3 paralogue Pax7 resulted in an axolotl phenotype that was similar to those seen in Pax3−/− and Pax7−/− mutant mice. The axolotl genome provides a rich biological resource for developmental and evolutionary studies.
Similar content being viewed by others


Main
Salamanders boast an illustrious history in biological research as the animal in which the Spemann organizer1 and Sperry’s chemoaffinity theory of axonal guidance2 were discovered. Since 1768, when Spallanzani discovered tail and limb regeneration, researchers have probed this animal’s remarkable regenerative capabilities with increasing molecular resolution. A. mexicanum (Fig. 1a) was first collected by von Humboldt, and has been cultivated in the laboratory since 1864 as a model for investigating phenomena such as nuclear reprogramming, the embryology of germ-cell induction, retinal neuron processing and regeneration3. Owing to the ease with which A. mexicanum can be bred in the laboratory, a sophisticated molecular toolkit has been developed for this species, including germline transgenesis and CRISPR-mediated gene mutation as well as viral and other transfection methods. These tools have enabled discoveries such as the identification of the source cells of regeneration and molecular pathways that control regeneration4,5. A full exploitation of the axolotl model, including understanding regeneration and why it is limited in other tetrapods, requires analysis of its genome regulation and evolution. However, efforts towards comprehensive assembly of salamander genomes have been challenging owing to their large genome sizes (14–120?Gb) and the large number of repetitive regions they contain; the 32-Gb axolotl genome is ten times the size of the human genome. Here we report the sequencing, assembly and analysis of the axolotl genome.

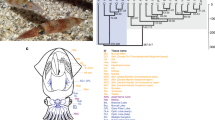
a, A wild-type A. mexicanum and the sequenced d/d A. mexicanum strain. b, The assembly strategy combines long-read sequencing, a novel assembler (MARVEL), error correction and scaffolding. c, A 57,385-bp PacBio read (red line) spans a large repetitive region (repeats are shown in orange; the longest repeat is 34?kb) and, together with the other long reads shown below the long PacBio read, allows assembly of the locus (green-to-red colouring indicates alignment quality; repeat-induced alignments of reads belonging to other loci have been removed). d, N(x) plot shows the percentage of the genome (x axis) that consists of contigs of at least x?kb (y axis).
A long-read assembler for large genomes
Our aim was to generate a genome sequence assembly for the d/d axolotl strain (Fig. 1a), which is commonly used in laboratory regeneration studies owing to its compatibility with live imaging. Taking into consideration the expected challenge of assembling the complex 32-Gb genome6, we sequenced 110?million long reads (32× coverage, N50 read length 14.2?kb) using Pacific Biosciences (PacBio) instruments (Supplementary Information section 1) to avoid the read sampling bias that is often found when using other technologies and to span repeat-rich genomic regions that cause breaks in short-read assemblies (Fig. 1b, c).
We developed an assembly algorithm (MARVEL) that integrates a two-phase read-correction procedure that keeps long PacBio reads intact for assembly (Supplementary Information section 2). MARVEL produced a contig assembly with an N50 of 218?kb. Next, we used 7× Illumina-based sequencing to correct sequence errors in 1% of the contig bases (Fig. 1b), which yielded a sequence accuracy of more than 99.2%. On the basis of the Illumina data, we estimated a heterozygosity of 0.47% (Supplementary Information section 2.2).
To provide a scaffold for the contig assembly, we generated de novo optical maps using the Bionano Saphyr system (Supplementary Information section 2.3). The Bionano map resolved contig chimaeras, which were found in 1.7% of contigs, slightly reducing N50 contig length to 216?kb (Fig. 1d). The final hybrid assembly yielded an N50 scaffold length of 3?Mb. Compared to the short-read assembly of the 20-Gb spruce genome7 or the 22-Gb loblolly pine genome8, which involved 12× long-read coverage, the axolotl assembly showed 56- and 29-fold improvements in contiguity, respectively (Table 1).
To assess the completeness of the assembly (Supplementary Information section 4.1), we first determined the number of aligning non-exonic ultraconserved elements9 (UCEs). We found that 194 (98.5%) of 197 non-exonic UCEs that are conserved across vertebrates align to the axolotl assembly. By comparison, 189 and 192 UCEs align to the Tibetan frog and Xenopus genomes, respectively, and 195 UCEs align to the coelacanth genome, indicating that the completeness of the axolotl genome assembly is comparable to or better than the two other amphibian genomes, which are smaller than 2.3?Gb10.
To further assess the completeness of the assembly, we generated a comprehensive gene catalogue by sequencing mRNA from 22 tissues (Supplementary Information section 3). Tissue-specific transcriptome assemblies and a composite assembly of all 1.5 billion transcript reads resulted in 180,649 transcript contigs (Supplementary Table 6) that contained 99% of the conserved core eukaryotic genes11 and achieved the highest BUSCO score (http://busco.ezlab.org/) of an axolotl transcriptome reported to date (Supplementary Information section 3.4). More than 85% of the transcripts aligned to the genome along at least 95% of their length (Supplementary Information section 3.5), confirming the high completeness of the assembly. Furthermore, 71% of transcript contigs in which more than 95% of the sequence aligned with the genome were located on single scaffolds, demonstrating the high contiguity of the assembly. Using this comprehensive transcript set, we annotated a total of 23,251 protein-coding genes in the axolotl genome, a similar number to those found in other vertebrate genomes (Supplementary Information section 4.2).
Expansion of long terminal repeat retroelements
Given the similar number of genes in the A. mexicanum genome in comparison to other smaller vertebrate genomes, we analysed repetitive sequences (Supplementary Information sections 4.2.2, 4.2.3). Repetitive sequences made up 65.6% of the contig assembly, representing a total of 18.6?Gb. Distinct long terminal repeat (LTR) retroelement classes and endogenous retroviruses made up the largest portion of the repetitive sequences (Fig. 2a, b, Supplementary Table 13) and included elements of more than 10?kb in length (Fig. 2c, Extended Data Fig. 1). Such long elements pose challenges for assembly, and indeed 97% of contigs ended in LTR elements. The number of substitutions to the repeat consensus, which is an estimate of the relative age of the LTR retroelement, indicates that the axolotl genome has undergone a long period of transposon activity followed by a recent and apparently continuing burst of expansion (Fig. 2d). This profile is consistent with previous small-scale characterizations of other salamander genomes12.

a, Pie charts of major repeat classes (LINE, long interspersed nuclear elements; SINE, short interspersed nuclear elements) show an abundance of LTR elements. b, Phylogenetic tree of axolotl LTR-element clusters (black) and all LTR elements from GyDB2.037. Annotated clusters are indicated by colour, non-annotated clusters are in grey. Note that Errantiviridae (blue), Caulimoviridae (red) and Athila/Tat (orange) families are not found. c, Box plots show the length distribution of LTR families (ERVL, endogenous retrovirus-like). Boxes indicate the first quartile, the median and the third quartile with whiskers extending up to 1.5 times the interquartile distance. Outliers are defined as data points outside the whiskers and are shown as dots. Quantitative data and sample sizes are shown in Source Data. d, Relative age (Kimura distance) suggests prolonged transposition activity followed by a recent activity burst.
The presence of many repeated elements contributes to a median intron size (22,759 bp) 13, 16 and 25 times that observed in human (1,750 bp), mouse (1,469 bp) and frog (906 bp), respectively (Fig. 3a, Supplementary Information section 4.3), a trend that was previously observed in five genes obtained from selective bacterial artificial chromosome sequencing of the axolotl genome13. Figure 3b shows a typical gene organization in axolotl compared to its human orthologue. Consistent with intron expansion, a distance comparison of pairs of highly conserved non-exonic elements shows that intergenic regions in the axolotl genome are 12 to 17 times larger than those in human, mouse and frog (Supplementary Information section 4.4).

a, Intron size of developmental genes appears to be under constraint in A. mexicanum. Violin plots represent the full distribution of intron sizes (thick bar, first to third quartile; white dot, median). ***P?<?10−11; two-sided Wilcoxon rank-sum tests. Quantitative data are shown in Source Data. b, Organization of the Agr2 exon–intron structure shows a consistent expansion of axolotl intron sizes compared to those in the human orthologue, resulting in a gene that is 4.3 times larger. c, Comparison of genes and repetitive elements in the HoxA cluster. CNEs that align to the axolotl HoxA cluster are shown in red. d, Axolotl lacks the Pax3 locus. Analysis of tetrapod-conserved genes and CNEs associated with Pax7 and Pax3 genomic loci. Red, gene sequences and CNEs that are absent in axolotl; *CNEs that overlap well-characterized mouse Pax3 enhancers38,39.
HoxA cluster and intron size constraints
To examine gene cluster organization within this large genomic context, we focused on the HoxA locus, which has an important role in proximal-to-distal limb development and is reactivated during limb regeneration14,15. The entire HoxA locus is contained on a single contig (Fig. 3c), and the conserved neighbouring gene Evx1 is contained on the same 3.34-Mb scaffold. Compared to the orthologous human and frog clusters, the A. mexicanum HoxA cluster has a substantially increased repeat content and is 3.5 times larger, mostly owing to a 170-kb expansion between HoxA3 and HoxA4 (Fig. 3c). Notably, highly conserved non-exonic elements that putatively overlap cis-regulatory elements are not interspersed in this 170-kb region, but remain in proximity to HoxA3 and HoxA4. The axolotl has a typical HoxA gene structure, with two coding exons separated by an intron. Notably, in contrast to the overall expansion of intron sizes, the intron sizes in the axolotl HoxA locus are very similar to those in other vertebrates, with the exception of AmHoxA3, which is also the longest of the HoxA genes in other tetrapods (Supplementary Table 17). Selected HoxC and HoxD genes examined in the red spotted newt exhibited similar properties16.
On the basis of these observations, we examined the intron size distribution among a larger set of orthologous genes involved in developmental processes. While introns of non-developmental genes in axolotl show a median size expansion of 13- to 25-fold compared to human, mouse and frog, the expansion of introns of developmental genes is significantly lower (6- to 11-fold, P?<?10−11) (Fig. 3a, Supplementary Information section 4.3). In contrast to human, mouse and frog, introns of developmental genes in axolotl are shorter than introns of non-developmental genes. Furthermore, axolotl multi-exon genes that contain only short introns exhibit gene ontology enrichments related to developmental patterning that are not enriched in multi-exon genes with larger introns (Supplementary Table 16). These results suggest that intron size in developmental genes is under constraint in the axolotl, possibly because smaller gene sizes facilitate rapid transcription and thus upregulation of these genes in specific developmental contexts.
A reduced Pax-family complement
Next, we interrogated the genome for families of canonical developmental signalling molecules (Supplementary Information section 5). All three hedgehog paralogues as well as a full set of vertebrate Wnt genes were present (Extended Data Fig. 2a, b). However, we noted that certain members of the paired box family of transcription factors, which have diverse roles in tissue formation, were not found in the assembly. Consistent with the absence of Pax4 in amphibians and other vertebrate lineages17, the axolotl genome does not contain Pax4 but does contain Pax10. Notably, despite the presence of the Pax3 and Pax7 paralogues in all other known vertebrate lineages, we were able to identify Pax7 but not Pax3 in the axolotl genome assembly (Extended Data Fig. 2c). No Pax3 sequence was found in either the raw PacBio sequencing reads or the transcriptome. To confirm the loss of Pax3, we further examined the genomic region that would be syntenic for Pax3 for the presence of neighbouring genes and highly-conserved non-exonic elements (CNEs). The orthologues of genes surrounding mouse Pax3 (Sgpp2 and Epha4) were present in the A. mexicanum genome assembly; however, neither the Pax3 gene nor any of the Pax3-associated CNEs were found (Fig. 3d). By contrast, several CNEs that overlap the Pax7 gene were identified in the assembly. Together, this evidence strongly suggests that Pax3 and several of its cis-regulatory elements are absent in the axolotl genome, probably owing to a deletion.
Axolotl Pax7 has similar functions to Pax3
To functionally assess the consequence of the absence of Pax3 in the axolotl, we used TALEN- and CRISPR-mediated gene editing18 to mutate Pax7. In other vertebrates, Pax3 and Pax7 play key roles in muscle, neural tube and neural crest-derived tissue development19. Although these two genes share some common functions, deletion of Pax3 or Pax7 causes distinct phenotypes in mice20,21,22. We investigated whether frameshift deletions introduced into the AmPax7 gene would yield a comparable Pax7 phenotype, or whether AmPax7 may have taken on functions that are carried out by Pax3 in other vertebrates. Two different TALEN-mutant alleles (7-nt and 20-nt deletions) of AmPax7 were bred through two generations (Fig. 4a, Supplementary Information section 6). In the F2 generation, the developmental phenotype described below was observed in 83 out of 345 (24%) progeny from the Pax7Δ20nt/+ intercrossing and 57 of 232 (24.6%) progeny from the Pax7Δ7nt/+ intercrossing (Fig. 4b, Extended Data Fig. 3). The phenotype was evident in homozygous mutants, as analysed by PCR and loss of protein (Supplementary Information sections 6.1, 6.3). This information, combined with the CRISPR-mediated gene mutation results (Supplementary Information sections 6.2), shows that the homozygous Pax7Δ20nt/Δ20nt and Pax7Δ7nt/Δ7nt mutants represent recessive, complete or partial loss of Pax7 function.

a, Deletion of AmPax7 coding sequences using TALEN and CRISPR. Deletions were made in exon 1 or exon 2. The first three AmPax7 exons (Ex) are shown. Red rectangles, TALEN targets; arrows, CRISPR-guide RNA (gRNA)-binding sites. b, Images of 6-month-old Pax7Δ20nt/Δ20nt mutants compared to controls show loss of body elongation. Scale bars, 1?cm. c, Reduced body wall muscle in Pax7 mutants. Immunofluorescence images of myosin heavy chain (MHC, red) and DAPI (blue) in trunk cross-sections from a 6-month-old Pax7Δ20nt/Δ20nt mutant (Pax7-TALEN#2) and a control animal. Scale bar, 500?μm. d, Limbs of Pax7 mutants lack muscle. Forelimb (left; scale bars, 500?μm) and immunofluorescence images of MHC (red) and DAPI (blue) in limb cross-sections (right; scale bars, 100?μm) of a 3-month-old Pax7Δ20nt/Δ20nt mutant and a control animal. e, Loss of prefrontal bone in Pax7 mutants. Dorsal and lateral views of Alcian blue and Alizarin red-stained Pax7Δ20nt/Δ20nt and Pax7-Ex1-CRISPR#3 mutants and controls (right). Red arrows, prefrontal bone; blue arrows, maxillary bone. The yellow arrowhead points to a small remnant of the prefrontal bone. Scale bar, 1?mm. f, g, Reduced melanophores, xanthophores (f) and iridophores (g) in Pax7 mutants. Images of 17-day-old Pax7-Ex2-CRISPR#1 (f), 2-month-old Pax7Δ20nt/Δ20nt (g) mutants and controls (Ctr). Right panels in f show a magnified view of the outlined area; red arrows in g point to the eyes that are magnified in the insets; black arrows indicate the belly iridophores in the control. Scale bars, 1?mm. h, Neural tube closure defect in Pax7 mutants. Images of a 31-day-old Pax7-Ex2-CRISPR#1mutant and control (Ctr). Right, magnified view of the outlined area. Scale bar, 1?mm. Quantitative data and sample sizes are provided in the Life Sciences Reporting Summary and Source Data.
The Pax7Δ20nt/Δ20nt and strong F0 Pax7-CRISPR mutants exhibited a curved body, were unable to maintain an erect posture and exhibited a delay in growth compared to controls. Immunohistochemical analysis of trunk or tail cross-sections of early stage, 20-day-old Pax7Δ20nt/Δ20nt or 17-day-old F0 Pax7-CRISPR axolotls showed normal muscle mass. However, at later stages, consistent with the mouse Pax7 deletion phenotype, tail and trunk muscles were greatly decreased (Fig. 4c, Extended Data Figs 4, 5, 6). Remarkably, the Pax7 mutant axolotls also completely lacked limb muscle (Figs 4d, Extended Data Fig. 7). In mice, Pax3, but not Pax7, is required for limb muscle formation21,22,23 (Supplementary Table 18). These results demonstrate that AmPax7 has comparable functions to MmPax3 in the control of limb muscle genesis.
In mice, Pax7 deletion affects craniofacial neural crest derivatives, including the facial bones20, whereas in zebrafish, pax7 mutants show loss of xanthophores and reduction of melanophores, but no loss of iridophores24. The AmPax7 mutants lacked a prefrontal bone, had a reduced number of melanophores and were deficient in xanthophores and iridophores except in the eyes (Fig. 4e–g, Extended Data Fig. 8). Pax3 deletion in mice is associated with neural tube closure defects22,23 (Supplementary Table 18). Similarly, Pax7Δ20nt/Δ20nt-TALEN and Pax7-CRISPR axolotls displayed failed closure of the neural tube in the midbrain (Fig. 4h, Extended Data Fig. 9). In summary, mutation of AmPax7 yields a combination of the Pax3- and Pax7-mutant phenotypes that are observed in other vertebrates (Supplementary Information section 6). It will be interesting to understand how the regulation of Pax7 has changed in axolotl to enable the loss of Pax3, which is essential in other vertebrates.
Species-restricted genes in regeneration
Previous searches for mRNA and microRNA (miRNA) transcripts associated with limb regeneration relied on mapping to de novo transcriptome assemblies. We sought to re-examine these datasets using our newly acquired genomic data. Recent functional work has highlighted the role of diverged gene or protein function during regeneration25,26,27. Analysis of published tissue-enriched datasets28, combined with regeneration time courses29,30 and our own transcriptional profiling of 22 tissues, identified five transcripts that are upregulated in the limb blastema (the mass of proliferating cells involved in regenerating the limb) with orthology limited to non-amniote vertebrates (Supplementary Information section 7). One of these protein sequences shows a weak similarity to tectorin, a basement membrane component normally found in the inner ear, consistent with studies that implicate extracellular matrix components with having an important role in limb regeneration31,32. Notably, another of these transcripts encodes a Ly6 family member in the urokinase type plasminogen activator surface receptor (uPAR) class. Previous studies had identified the salamander-specific Ly6 family member Prod1 as a key factor involved in salamander limb development and regeneration25,33. Our results suggest that Ly6 family members have a broader role in limb regeneration. Finally, we also investigated the role of non-coding RNAs by mapping a dataset of small RNA sequences expressed in the limb and limb blastema34 to our genome assembly. This analysis classified 93 small RNAs as pre-miRNA sequences, of which 42 appear to be novel miRNAs (Supplementary Information section 7.2). Taken together, these data point to a potential role in limb regeneration for several coding and non-coding sequences that have been lost or diverged rapidly in amniotes. Future investigations of such sequences are likely to be a fruitful avenue for understanding the evolution of regeneration capabilities.
Discussion
We have generated a comprehensive whole-genome assembly for the salamander A. mexicanum, and analysis of this assembly has allowed us to draw conclusions about the structure of the expanded genome. Our data, together with data from plants and partial data from several other salamander species, show that LTR expansion is a major contributor to giant genome size across animals and plants6,12,35. Our assembly is sufficiently complete to reliably detect the absence of Pax3, which is present in fish and other amphibians. This analysis was confirmed using gene editing, which showed that AmPax7 has assumed functions that are carried out by Pax3 in other animals.
Functional analysis of axolotl development, physiology and regeneration is facilitated by the availability of tissue- and time-dependent gene expression profiles28,29,30,36. The axolotl genome provides a foundation for applying methods such as chromatin immunoprecipitation with sequencing (ChIP–seq) or assay for transposase-accessible chromatin using sequencing (ATAC-seq) to investigate the genomic basis of gene regulation during regeneration. Together with methods such as CRISPR-mediated gene editing, viral expression methods, transplantation and transgenesis, the axolotl is a powerful system for studying questions such as the evolutionary basis of its remarkable regeneration ability. Our approach of long-read sequencing, optical mapping and genome assembly using MARVEL also demonstrates that it is now feasible to assemble very large repeat-rich genomes.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.Axolotl genomic DNA was prepared from freshly isolated liver and spleen of an individual three year old adult d/d male using DNAzol followed by phenol/chloroform extraction and ethanol precipitation.
A total of 50 size-selected SMRTbell libraries were prepared with a minimum fragment length cutoff between 10?kb and 20?kb. We sequenced medium and large insert libraries on the PacBio RSII instrument, making use of three different sequencing polymerases (P4, P5 and P6) and the corresponding sequencing chemistries (C2, C3 and C4). Movie times ranged from 180?min to 360?min with the majority of SMRT cells (1,414 of 1,933) at 240?min.
Sequences were assembled using the MARVEL assembler.
Optical mapping was performed using the Saphyr System (Bionano) based on NanoChannel array Technology. DNA was labelled with Nt.BspQI and Nb.BssSI enzymes in separate labelling reactions. Each enzyme reaction was run on the Saphyr System. 2.813 Tb of data were collected on three Saphyr Chips for Nt.BspQI and 2.0 Tb of data were collected on two Saphyr Chips for Nb.BssSI samples; single molecule N50 lengths were 240?kb and 184?kb, respectively. Each dataset was de novo assembled using Bionano Solve 2.1 software.
RNA was isolated from 22 tissue types using TRIzol or RNeasy reagents and sequenced using Illumina technology. The Trinity software package was used for transcriptome assembly.
Code availability
The MARVEL assembler with documentation is available at https://github.com/schloi/MARVEL.
Data availability
A browser of the axolotl genome is available at https://genome.axolotl-omics.org. The transcriptome assembly and the genome and transcriptome BLAST database can be accessed at https://www.axolotl-omics.org with no restrictions. The sequence data and both assemblies have been deposited in the NCBI BioProject database with accession numbers PRJNA378970 (genome data) and PRJNA378982 (transcriptome data). Both genome data and transcriptome data were deposited to the NCBI Nucleotide Database (nuccore) with accession numbers PGSH00000000 and GFZP00000000, respectively.
Accession codes
Primary accessions
BioProject
NCBI Reference Sequence
Change history
23 May 2018
In the originally published version of this Article, the axolotl strain was denoted as 'D/D' rather than 'd/d'; this has been corrected online.
References
Spemann, H. & Mangold, H. Über Induktion von Embryonalanlagen durch Implantation artfremder Organisatoren. Arch. Mikrosk. Anat. En. 100, 599–638 (1924)
Sperry, R. W. Effect of 180 degree rotation of the retinal field on visuomotor coordination. J. Exp. Zool. 92, 263–279 (1943)
Voss, S. R., Epperlein, H. H. & Tanaka, E. M. Ambystoma mexicanum, the axolotl: a versatile amphibian model for regeneration, development, and evolution studies. Cold Spring Harb. Protoc. 2009, https://doi.org/10.1101/pdb.emo128 (2009).
Currie, J. D. et al. Live imaging of axolotl digit regeneration reveals spatiotemporal choreography of diverse connective tissue progenitor pools. Dev. Cell 39, 411–423 (2016)
Tanaka, E. M. The molecular and cellular choreography of appendage regeneration. Cell 165, 1598–1608 (2016)
Keinath, M. C. et al. Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing. Sci. Rep. 5, 16413 (2015)
Warren, R. L. et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 83, 189–212 (2015)
Zimin, A. V. et al. An improved assembly of the loblolly pine mega-genome using long-read single-molecule sequencing. Gigascience 6, 1–4 (2017)
Bejerano, G. et al. Ultraconserved elements in the human genome. Science 304, 1321–1325 (2004)
Sun, Y.-B. et al. Whole-genome sequence of the Tibetan frog Nanorana parkeri and the comparative evolution of tetrapod genomes. Proc. Natl Acad. Sci. USA 112, E1257–E1262 (2015)
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007)
Sun, C. & Mueller, R. L. Hellbender genome sequences shed light on genomic expansion at the base of crown salamanders. Genome Biol. Evol. 6, 1818–1829 (2014)
Smith, J. J. et al. Genic regions of a large salamander genome contain long introns and novel genes. BMC Genomics 10, 19 (2009)
Davis, A. P., Witte, D. P., Hsieh-Li, H. M., Potter, S. S. & Capecchi, M. R. Absence of radius and ulna in mice lacking hoxa-11 and hoxd-11. Nature 375, 791–795 (1995)
Roensch, K., Tazaki, A., Chara, O. & Tanaka, E. M. Progressive specification rather than intercalation of segments during limb regeneration. Science 342, 1375–1379 (2013)
Voss, S. R. et al. Salamander Hox clusters contain repetitive DNA and expanded non-coding regions: a typical Hox structure for non-mammalian tetrapod vertebrates? Hum. Genomics 7, 9 (2013)
Feiner, N., Meyer, A. & Kuraku, S. Evolution of the vertebrate Pax4/6 class of genes with focus on its novel member, the Pax10 gene. Genome Biol. Evol. 6, 1635–1651 (2014)
Gaj, T., Gersbach, C. A. & Barbas, C. F. 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31, 397–405 (2013)
Mansouri, A., Hallonet, M. & Gruss, P. Pax genes and their roles in cell differentiation and development. Curr. Opin. Cell Biol. 8, 851–857 (1996)
Mansouri, A., Stoykova, A., Torres, M. & Gruss, P. Dysgenesis of cephalic neural crest derivatives in Pax7−/− mutant mice. Development 122, 831–838 (1996)
Kuang, S., Chargé, S. B., Seale, P., Huh, M. & Rudnicki, M. A. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J. Cell Biol. 172, 103–113 (2006)
Relaix, F., Rocancourt, D., Mansouri, A. & Buckingham, M. Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 18, 1088–1105 (2004)
Auerbach, R. Analysis of the developmental effects of a lethal mutation in the house mouse. J. Exp. Zool. 127, 305–329 (1954)
Nord, H., Dennhag, N., Muck, J. & von Hofsten, J. Pax7 is required for establishment of the xanthophore lineage in zebrafish embryos. Mol. Biol. Cell 27, 1853–1862 (2016)
da Silva, S. M., Gates, P. B. & Brockes, J. P. The newt ortholog of CD59 is implicated in proximodistal identity during amphibian limb regeneration. Dev. Cell 3, 547–555 (2002)
Garza-Garcia, A. A., Driscoll, P. C. & Brockes, J. P. Evidence for the local evolution of mechanisms underlying limb regeneration in salamanders. Integr. Comp. Biol. 50, 528–535 (2010)
Sugiura, T., Wang, H., Barsacchi, R., Simon, A. & Tanaka, E. M. MARCKS-like protein is an initiating molecule in axolotl appendage regeneration. Nature 531, 237–240 (2016)
Bryant, D. M. et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Reports 18, 762–776 (2017)
Stewart, R. et al. Comparative RNA-seq analysis in the unsequenced axolotl: the oncogene burst highlights early gene expression in the blastema. PLOS Comput. Biol. 9, e1002936 (2013)
Knapp, D. et al. Comparative transcriptional profiling of the axolotl limb identifies a tripartite regeneration-specific gene program. PLoS ONE 8, e61352 (2013)
Calve, S., Odelberg, S. J. & Simon, H.-G. A transitional extracellular matrix instructs cell behavior during muscle regeneration. Dev. Biol. 344, 259–271 (2010)
Tassava, R. A., Nace, J. D. & Wei, Y. Extracellular matrix protein turnover during salamander limb regeneration. Wound Repair Regen. 4, 75–81 (1996)
Kumar, A., Gates, P. B., Czarkwiani, A. & Brockes, J. P. An orphan gene is necessary for preaxial digit formation during salamander limb development. Nat. Commun. 6, 8684 (2015)
King, B. L. & Yin, V. P. A conserved microRNA regulatory circuit is differentially controlled during limb/appendage regeneration. PLoS ONE 11, e0157106 (2016)
Sun, C. et al. LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders. Genome Biol. Evol. 4, 168–183 (2012)
Jiang, P. et al. Analysis of embryonic development in the unsequenced axolotl: waves of transcriptomic upheaval and stability. Dev. Biol. 426, 143–154 (2017)
Llorens, C. et al. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 39, D70–D74 (2011)
Milewski, R. C. et al. Identification of minimal enhancer elements sufficient for Pax3 expression in neural crest and implication of Tead2 as a regulator of Pax3. Development 131, 829–837 (2004)
Degenhardt, K. R. et al. Distinct enhancers at the Pax3 locus can function redundantly to regulate neural tube and neural crest expressions. Dev. Biol. 339, 519–527 (2010)
Acknowledgements
E.W.M. was supported by the Max Planck Society President's fund and BMBF grants 01IS14014C and 031L0102. E.M.T., S.N. and J.-F.F. were supported by MPI-CBG, DFG FZ111 and IMP. E.M.T. and J.-F.F. were supported by DFG TA 274/3-3. J.-F.F. is supported by NSFC grant 31771611 and a Research Starting Grant S82111-E2 from SCNU. F.F. was supported by a CONACYT Masters Fellowship. A.C. was supported by The Swedish IRLG GRANT 2014-9040-114152-32, CONACYT FOINS-301 and Ciencia Básica I0017-CB-2015-01-000000000252126. G.Y. was supported by the Crick Institute award FC001162 (to J. P. Stoye). The Crick receives its core funding from CRUK, the MRC and the Wellcome Trust. B.H. was supported by BMBF grant 01IH11003C. We thank W. Bonacci, E. Gromberg, A. Tazaki and I. Stützer for sample preparation; S. Clausing, N. Gscheidel, Y. Duport, A. Sommer and the VBCF NGS facility for sequencing; A. Kavirayani and T. Engelmaier at VBCF for histology; E. Heude for confirmation of Pax7 mutant immunofluorescence; T. Anantharaman for optimizing assembly and scaffolding parameters for Bionano data; and G. Papoutsoglou for advice.
Author information
Authors and Affiliations
Contributions
S.N. assembled and analysed the transcriptome, and performed genome analysis. S.S. created, engineered and implemented the assembly algorithm and analysed the genome assembly. J.-F.F. performed DNA extraction and all biological experiments and analysis. M.P. and S.P. contributed to implementing the assembler, assembled the genome and contributed to the genome analysis. A.D. and S.W. performed Pacific Biosciences sequencing. G.Y. analysed the LTR elements. J.G.R. performed the conserved element and intron analysis. F.F. and A.C. performed the developmental orthologue and miRNA analysis. A.W.C.P., A.R.H. and H.C. performed Bionano optical mapping, generated the hybrid and scaffolded the assembly. D.K. performed tectorin analysis. B.H. supervised transcriptome assembly and annotation and acquired funding. E.M.T. and M.H. conceived analytical strategies, performed data analysis and acquired funding. S.N., S.S., J.-F.F., M.P., S.P., E.M.T. and M.H. wrote the manuscript. E.W.M. conceived and implemented the assembly strategy, acquired major funding and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks C. Crews and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Analysis of LTR retroelement frequencies according to their lengths.
The line shows a moving average (period 25) to highlight clusters of elements of similar lengths.
Extended Data Figure 2 Phylogenetic trees.
a, Phylogenetic tree of vertebrate hedgehog proteins show the presence of axolotl orthologues. b, Phylogenetic tree of vertebrate Wnt proteins show the presence of axolotl orthologues in all Wnt classes. c, Phylogenetic tree of vertebrate PAX proteins. Pax4 and Pax3 are absent in axolotl.
Extended Data Figure 3 Developmental phenotype of Pax7 mutants.
a, b, Images of live Pax7Δ20nt/Δ20nt mutants compared to controls show no obvious phenotype at early stages (a, 1 month), but an obvious phenotype at later stages (b, 3.5 months). c, d, Images of live F0 Pax7-Ex1-CRISPR#3 (c, 4 months) and Pax7-Ex2-CRISPR#1 (d, 6 months) mutants show the curved body phenotype. Scale bars, 1?cm. Numbers of replicate matings and experiments are shown in the Life Sciences Reporting Summary and Source Data.
Extended Data Figure 4 Progressive depletion of the trunk muscle in Pax7 mutants.
a, Images of live Pax7Δ20nt/Δ20nt animals at different ages compared to corresponding controls show the progressive loss of the trunk muscle in mutant animals. Black arrows indicate trunk muscles; blue arrows highlight the visibility of the spine after reduction and/or depletion of trunk muscle. Scale bars, 2mm. b, c, Images of live 7-month-old F0 Pax7-Ex1-CRISPR#3 (b) and 6-month-old F0 Pax7-Ex2-CRISPR#1 (c) mutants compared to controls, showing loss of trunk muscle. Black arrows indicate trunk muscles; blue arrows indicate the visibility of the spine after depletion of trunk muscle. Scale bars, 5?mm. Number of replicate matings and experiments are shown in the Life Sciences Reporting Summary and Source Data.
Extended Data Figure 5 Progressive depletion of the tail muscle in Pax7Δ20nt/Δ20nt mutants.
a, b, Images of live 75-day (a) and 6-month-old (b) Pax7Δ20nt/Δ20nt homozygous mutants compared to controls show the progressive depletion of tail muscle. White arrows indicate tail muscle fibres; right, magnified view of the outlined area. Note the decrease in myotome length in 75-day-old Pax7Δ20nt/Δ20nt homozygous mutants (b). Scale bars, 500?μm. c, Immunofluorescence images of MHC (red) and DAPI (blue) in tail cross-sections show reduction in tail muscle in 3-month-old Pax7Δ20nt/Δ20nt mutants compared to controls. Scale bar, 100?μm. Number of replicate matings and experiments are shown in the Life Sciences Reporting Summary and Source Data.
Extended Data Figure 6 Depletion of the abdominal muscle in Pax7 mutants.
a, b, Bright-field image and GFP fluorescence of live 34-day (a) and 64-day-old (b) F0 Pax7-Ex1-CRISPR#3 mutants obtained by injecting Pax7-gRNA#3–CAS9 protein complex into eggs of CarAct:EGFP transgenic axolotls, compared to un-injected CarAct:EGFP controls. Mutant animals show a reduction in the EGFP-labelled abdominal muscles. Right, magnified view of GFP fluorescence in the outlined area; white arrows indicate forelimbs that either contain or lack EGFP-labelled muscles; green arrows indicate the GFP-labelled abdominal muscle; red arrows indicate regions that lack GFP-labelled abdominal muscle. Scale bars, 1 mm. c, Images of live 6-month-old Pax7Δ20nt/Δ20nt mutants compared to controls show the loss of abdominal muscle. Scale bars, 1?mm. d, Immunofluorescence images of MHC (red) and DAPI (blue) in cross-sections show the presence of ventral body-wall muscle in the chest position in 6-month-old Pax7Δ20nt/Δ20nt homozygous mutants compared to controls, and the gradual depletion of the abdominal muscle along the anterior–posterior axis. Arrows, skin; yellow stars, liver; green stars, intestine. Scale bar, 100?μm. Number of replicate matings and experiments are shown in the Life Sciences Reporting Summary and Source Data.
Extended Data Figure 7 Loss of limb muscle in Pax7 mutants.
a, Images of live 54-day-old Pax7Δ20nt/Δ20nt mutants compared to controls show loss of limb muscle. Right, magnified view of the outlined area. Scale bars, 1?mm. b, c, Non-muscle tissues are normal in Pax7Δ20nt/Δ20nt mutant limbs. Immunofluorescence images for TU-J1 (b, green) MBP (b, red), CO1A2 (c, green) and DAPI (blue) in forelimb cross-sections of 56-day-old Pax7Δ20nt/Δ20nt mutants and controls. Scale bars, 100?μm. d, Images of live 80-day-old F0 Pax7-Ex1-CRISPR#3 heterozygotes compared to controls show loss of forelimb (FL) and hindlimb (HL) muscle on one side of the body (green arrows) but not on the other side (red arrows). Scale bar, 1?mm. e, Images of live 54-day-old F0 Pax7-Ex2-CRISPR#1 and Pax7-Ex2-CRISPR#3 mutants compared to a control (bottom) showing loss of forelimb muscle in CRISPR animals. Right, magnified view of the outlined area. Scale bars, 1 mm. Number of replicate matings and experiments are shown in the Life Sciences Reporting Summary and Source Data.
Extended Data Figure 8 Reduced melanophores, loss of xanthophores and iridophores in Pax7 mutants.
a, Images of a live 25-day-old Pax7Δ20nt/Δ20nt homozygous mutant compared to a control animal showing loss of xanthophores and reduction of melanophores in the head and neck region of mutant animals. Right, magnified view of the outlined area. Scale bar, 1?mm. b, Images of a live 54-day-old Pax7Δ20nt/Δ20nt homozygous mutant compared to a control animal showing a reduction in melanophores along the body. Arrows, melanophores. Scale bar, 500?μm. c. Images of a live 17-day-old F0 Pax7-Ex2-CRISPR#3 mutant compared to a control animal showing loss of xanthophores and reduction in melanophores in the head and neck region. Right, magnified view of the outlined area. Scale bar, 1?mm. d, Images of a live 2-month-old Pax7Δ20nt/Δ20nt homozygous mutant compared to a control animal showing loss of iridophores on the belly. Red arrows point to the eye, which is displayed at higher magnification showing eye pigmentation defects; green arrows indicate the presence of iridophores in the control animal (with silver eyes), but not in the mutant (with black eyes). Iridophores are absent in the Pax7-TALEN#2 mutants, irrespective of the eye colour. Scale bars, 1?mm. e, Images of live 6-month-old Pax7-Ex2-CRISPR#1 and Pax7-Ex2-CRISPR#3 mutants compared to a control animal, showing the reduction or loss of belly iridophores (right) in axolotls with silver eyes (left). Red arrows point to the eye, which is displayed at a higher magnification on the right; green arrows indicate remaining iridophores in F0 mosaic Pax7-CRISPR mutants or iridophores in the control animal. Scale bars, 1?mm. Number of replicate matings and experiments are shown in the Life Sciences Reporting Summary and Source Data.
Extended Data Figure 9 Neural tube closure defects in Pax7 mutants.
a, Images of a live 55-day-old Pax7Δ20nt/Δ20nt mutant compared to a control animal show an open brain phenotype. Right, magnified view of the outlined area. Scale bar, 1?mm. b–d, Images of live 15-day (b), 31-day (c) and 6-month-old (d) Pax7-Ex2-CRISPR#1 and Pax7-Ex2-CRISPR#3 mutants compared to controls, showing an open brain phenotype. Right, magnified view of the outlined area. Scale bar, 1?mm. e, Haematoxylin and eosin-stained paraffin cross-sections show the open neural tube of a 17-day-old F0 Pax7-Ex2-CRISPR#1 mutant compared to a control. Red arrows indicate the boundaries of the opened neural tube (NT). Scale bar, 200 μm. Number of replicate matings and experiments are shown in the Life Sciences Reporting Summary and Source Data.
Supplementary information
Supplementary Information
This file contains Supplementary Methods, Supplementary Figures 1-19, Supplementary Tables 1-21, Supplementary Data and additional references. (PDF 13050 kb)
Supplementary Figures
This file contains raw images for Supplementary Figure 17. (PDF 1263 kb)
Supplementary Table 22
This file contains the distance (bp) between CNE pairs in axolotl, Xenopus, human and mouse. (XLSX 93 kb)
Supplementary Table 23
This file contains members of the SHH, Wnt, and PAX protein families. (XLSX 46 kb)
Supplementary Table 24
This file contains axolotl homologs of the members of the SHH, Wnt, and PAX protein families. (XLSX 80 kb)
Supplementary Table 25
This file contains an expression of up-regulated transcripts in different tissues. (XLSX 39 kb)
Supplementary Table 26
This file contains a list of microRNAs. (XLSX 82 kb)
Supplementary Data
This file contains source data for Supplementary Table 16. (XLSX 257 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Nowoshilow, S., Schloissnig, S., Fei, JF. et al. The axolotl genome and the evolution of key tissue formation regulators. Nature 554, 50–55 (2018). https://doi.org/10.1038/nature25458
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25458
This article is cited by
-
Genomic reconsideration of fish non-monophyly: why cannot we simply call them all ‘fish’?
Ichthyological Research (2024)
-
Conserved chromatin and repetitive patterns reveal slow genome evolution in frogs
Nature Communications (2024)
-
Regulation of chromatin organization during animal regeneration
Cell Regeneration (2023)
-
The Information Theory of Aging
Nature Aging (2023)
-
A scATAC-seq atlas of chromatin accessibility in axolotl brain regions
Scientific Data (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
