
Abstract
The challenges of evolution in a complex biochemical environment, coupling genotype to phenotype and protecting the genetic material, are solved elegantly in biological systems by the encapsulation of nucleic acids. In the simplest examples, viruses use capsids to surround their genomes. Although these naturally occurring systems have been modified to change their tropism1 and to display proteins or peptides2,3,4, billions of years of evolution have favoured efficiency at the expense of modularity, making viral capsids difficult to engineer. Synthetic systems composed of non-viral proteins could provide a ‘blank slate’ to evolve desired properties for drug delivery and other biomedical applications, while avoiding the safety risks and engineering challenges associated with viruses. Here we create synthetic nucleocapsids, which are computationally designed icosahedral protein assemblies5,6 with positively charged inner surfaces that can package their own full-length mRNA genomes. We explore the ability of these nucleocapsids to evolve virus-like properties by generating diversified populations using Escherichia coli as an expression host. Several generations of evolution resulted in markedly improved genome packaging (more than 133-fold), stability in blood (from less than 3.7% to 71% of packaged RNA protected after 6 hours of treatment), and in vivo circulation time (from less than 5 minutes to approximately 4.5 hours). The resulting synthetic nucleocapsids package one full-length RNA genome for every 11 icosahedral assemblies, similar to the best recombinant adeno-associated virus vectors7,8. Our results show that there are simple evolutionary paths through which protein assemblies can acquire virus-like genome packaging and protection. Considerable effort has been directed at ‘top-down’ modification of viruses to be safe and effective for drug delivery and vaccine applications1,9,10; the ability to design synthetic nanomaterials computationally and to optimize them through evolution now enables a complementary ‘bottom-up’ approach with considerable advantages in programmability and control.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Deverman, B. E. et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209 (2016)
Chackerian, B., Caldeira, Jdo, C., Peabody, J. & Peabody, D. S. Peptide epitope identification by affinity selection on bacteriophage MS2 virus-like particles. J. Mol. Biol. 409, 225–237 (2011)
Smith, G. P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317 (1985)
Söderlind, E., Simonsson, A. C. & Borrebaeck, C. A. Phage display technology in antibody engineering: design of phagemid vectors and in vitro maturation systems. Immunol. Rev. 130, 109–124 (1992)
Bale, J. B. et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 353, 389–394 (2016)
Hsia, Y. et al. Design of a hyperstable 60-subunit protein icosahedron. Nature 535, 136–139 (2016)
Drouin, L. M. et al. Cryo-electron microscopy reconstruction and stability studies of the wild type and the R432A variant of adeno-associated virus type 2 reveal that capsid structural stability is a major factor in genome packaging. J. Virol. 90, 8542–8551 (2016)
Sommer, J. M. et al. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol. Ther. 7, 122–128 (2003)
Pascual, E. et al. Structural basis for the development of avian virus capsids that display influenza virus proteins and induce protective immunity. J. Virol. 89, 2563–2574 (2015)
Waehler, R., Russell, S. J. & Curiel, D. T. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 8, 573–587 (2007)
Harrison, S. C., Olson, A. J., Schutt, C. E., Winkler, F. K. & Bricogne, G. Tomato bushy stunt virus at 2.9 Å resolution. Nature 276, 368–373 (1978)
Lilavivat, S., Sardar, D., Jana, S., Thomas, G. C. & Woycechowsky, K. J. In vivo encapsulation of nucleic acids using an engineered nonviral protein capsid. J. Am. Chem. Soc. 134, 13152–13155 (2012)
Hernandez-Garcia, A. et al. Design and self-assembly of simple coat proteins for artificial viruses. Nat. Nanotechnol. 9, 698–702 (2014)
Wörsdörfer, B., Woycechowsky, K. J. & Hilvert, D. Directed evolution of a protein container. Science 331, 589–592 (2011)
Puglisi, J. D., Chen, L., Blanchard, S. & Frankel, A. D. Solution structure of a bovine immunodeficiency virus Tat-TAR peptide-RNA complex. Science 270, 1200–1203 (1995)
Starita, L. M. & Fields, S. Deep mutational scanning: a highly parallel method to measure the effects of mutation on protein function. Cold Spring Harb. Protoc. 2015, 711–714 (2015)
Whitehead, T. A. et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 30, 543–548 (2012)
Knop, K., Hoogenboom, R., Fischer, D. & Schubert, U. S. Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew. Chem. Int. Edn Engl. 49, 6288–6308 (2010)
Hui, D.J. et al. AAV capsid CD8+ T-cell epitopes are highly conserved across AAV serotypes. Mol. Ther. Methods Clin. Dev. 2, 15029 (2015)
Mingozzi, F. et al. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 13, 419–422 (2007)
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009)
Kunkel, T. A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl Acad. Sci. USA 82, 488–492 (1985)
Rohland, N. & Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22, 939–946 (2012)
Alvarez, P., Buscaglia, C. A. & Campetella, O. Improving protein pharmacokinetics by genetic fusion to simple amino acid sequences. J. Biol. Chem. 279, 3375–3381 (2004)
Schellenberger, V. et al. A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 27, 1186–1190 (2009)
Benson, D. A. et al. GenBank. Nucleic Acids Res. 41, D36–D42 (2013)
Nannenga, B. L., Iadanza, M. G., Vollmar, B. S. & Gonen, T. Overview of electron crystallography of membrane proteins: crystallization and screening strategies using negative stain electron microscopy. Curr. Protoc. Protein Sci. Chapter 17, Unit17.15 (2013)
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005)
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007)
Fowler, D. M., Araya, C. L., Gerard, W. & Fields, S. Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics 27, 3430–3431 (2011)
Hunter, J. D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007)
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015)
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009)
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protocols 11, 1650–1667 (2016)
Acknowledgements
We thank R. Chari for RNA-seq advice; S. Bustin for RT–qPCR advice; E. Gray and N. Arroyo for heparinized mouse blood; D. Veesler, J. Kollman and M. Johnson for EM advice; Y. Hsia for DLS advice; C. Walkey, Y. Hsia, G. Rocklin, J. Nelson, A. Chatterjee, S. Kosuri, G. Church, J. Bloom and A. Hessel for suggestions. This work was supported by the Howard Hughes Medical Institute (D.B.), the Bill and Melinda Gates Foundation (D.B. and N.P.K., grant number OPP1118840), the Defense Advanced Research Projects Agency (D.B. and N.P.K., grant number W911NF-15-1-0645), and the NIH (S.H.P., grant number NIH1R01CA177272; D.L.S., grant number NIH1R21NS099654-01A1). G.L.B. was supported by a National Science Foundation Graduate Fellowship. M.J.L. was supported by a Washington Research Foundation Innovation Postdoctoral Fellowship and a Cancer Research Institute Irvington Fellowship from the Cancer Research Institute. H.H.G. was supported by an NIH training grant (NIH5T32HL0071312). U.N. was supported in part by a PHS National Research Service Award (T32GM007270) from NIGMS.
Author information
Authors and Affiliations
Contributions
G.L.B. and M.J.L. designed the research and the experimental approach with guidance from N.P.K. and D.B.; G.L.B. and M.J.L. performed the evolution, nucleocapsid characterization, Illumina sequencing, and data analysis; H.H.G. and D.L.S. designed and performed the in vivo mouse experiments, and samples were processed by G.L.B. and M.J.L.; U.N. designed, performed, and analysed electron microscopy experiments; D.E. and J.B.B. designed the starting protein assemblies that were subsequently used for RNA packaging; S.K., G.H.L., A.Y. and R.R. assisted with cloning and protein purification; S.H.P., N.P.K. and D.B. supervised the research; G.L.B. and M.J.L. wrote the manuscript and produced the figures with guidance from H.H.G., D.L.S., U.N., S.H.P., N.P.K. and D.B.; G.L.B., M.J.L., H.H.G., D.L.S., U.N., J.B.B., S.H.P., N.P.K. and D.B. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks D. Schaffer and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 I53-47 nucleocapsids and SEC.
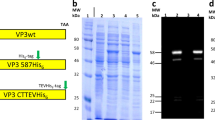
a, Design model of I53-47 and negative-stain electron micrographs of I53-47-v1 (designed positively charged interior) and I53-47-Btat (BIV Tat RNA-binding peptide translationally fused to the C terminus of the capsid trimeric subunit). Micrographs shown are representative of the entire sample tested on between one and three different grids, each at different concentrations. b, Synthetic nucleocapsids were Ni-NTA-purified, RNase-treated, and electrophoresed on non-denaturing 1% agarose gels. The gels were stained with Coomassie (protein; b) and SYBR gold (nucleic acid, c). Nucleic acids co-migrated with capsid proteins for all three versions of I53-47, suggesting that all versions package nucleic acid. d, Full-length synthetic nucleocapsid genomes were recovered from each sample by RT–qPCR. Plus and minus symbols indicate PCR performed on templates prepared with and without reverse transcriptase, respectively, confirming that all versions package their own full-length RNA genomes. This procedure is part of our standard quality control for synthetic nucleocapsids and has been performed reproducibly more than 10 times, including once on the I53-47 nucleocapsid shown here. e–h, SEC of nucleocapsids. RNA-packaging capsids show identical SEC retention volume as the original published capsid5. Three versions of I53-50 and I53-47 were analysed: v0 is the original published design, v1 has the designed positively charged interior, and Btat has the BIV Tat RNA-binding peptide translationally fused to the C terminus of the capsid trimer subunit. e, SEC traces of I53-50 capsids were performed on a GE superose 6 increase column. f, SDS–PAGE of samples before and after SEC purification shows both subunits in the expected 1:1 stoichiometry. g, h, SEC traces and SDS–PAGE for I53-47 capsids. This procedure is part of our standard quality control for synthetic nucleocapsids and has been performed reproducibly more than 10 times.
Extended Data Figure 2 Top synthetic nucleocapsid candidates for I53-50-v2 and I53-50-v3.
a, Top candidate testing to choose I53-50-v2 with improved genome packaging. New variants were created rationally based on the best sequences from the evolved interior charge optimization (Fig. 2) and interface (Supplementary Fig. 2) libraries. The amount of packaged full-length mRNA was compared for each of these nucleocapsids. Each nucleocapsid was expressed, purified by IMAC, and treated with 10 μg ml−1 RNase A at 20 °C for 10 min in triplicate. RT–qPCR was used to determine the relative amount of full-length mRNA packaged in each variant. Cq values are reported relative to those of I53-50-v1 (CqI53-50-v1 − Cqvariant). The charge-optimized variant with E24F was chosen as I53-50-v2 based on these data. In the absence of a discernable difference in packaging between E24M and E24F, E24F was selected owing to the apparent preference for hydrophobic residues at that position (Supplementary Fig. 2). Data points represent the values of three independent biological replicates, and error bars represent s.e.m. b, c, Top candidate testing to choose I53-50-v3 with improved nuclease resistance. b. Heat map of log enrichments for each mutation explored in the combinatorial library to remove positively charged residues near the nucleocapsid pore. A single round of selection (10 μg ml−1 RNase A, 37 °C, 1 h) was performed. Purple and orange indicate mutations that were depleted or enriched in the selected population, respectively. Blue squares and black dots indicate the I53-50-v2 starting sequence and I53-50-v3 selected sequence, respectively. c, Enriched variants selected from the combinatorial library were expressed, purified by IMAC and SEC, and treated with 10 μg ml−1 RNase A at 37 °C for 1 h in duplicate. RT–qPCR was used to determine the relative amount of full-length mRNA packaged in each variant. Cq values are reported relative to those of I53-50-v2 (CqI53-50-v2 − Cqvariant). Data points represent the values of two independent biological replicates, and bars represent the mean of these values. The variant labelled Pore_Mut_4 was chosen as I53-50-v3 based on this data.
Extended Data Figure 3 Evolution and performance of nucleocapsids modified with hydrophilic polypeptides in vitro or in vivo.
a, The change in population fraction corresponding to each variant was calculated from Illumina MiSeq counts for the input pool (t = 0), RNA recovered from circulation after 30 min (n = 3 biologically independent mice), and RNA recovered from circulation after 60 min (n = 2 biologically independent mice). b, Scatter plot of log10 enrichment of each hydrophilic polypeptide versus its net charge as calculated from the total number of charged residues in its sequence. c, Scatter plot of log10 enrichment of each polypeptide versus the number of unique amino acids in its sequence. d, Each of 11 variants were individually expressed and purified by IMAC before being pooled (equal protein concentration) and purified en masse by SEC. The resulting nucleocapsid pool was then incubated in heparinized whole blood at 37 °C (n = 3 independent reactions per time point). RNA was recovered at the indicated time points, and the fraction of each variant was determined by Illumina MiSeq counts taken at each time point. e. The same nucleocapsid pool used in d was injected retro-orbitally into mice (n = 5 biologically independent mice). RNA content was then assessed as in d using RNA isolated from tail vein draws at the indicated time points. All variants exhibit high stability in blood; however, the unmodified I53-50-v3 nucleocapsid (no polypeptide, blue) and a negative control polypeptide (ESESG, red) are cleared rapidly from circulation in vivo. Error bars represent s.e.m. The lower error bar for the pink data point at 15 min is not shown because its s.e.m. is nearly equivalent to its value.
Extended Data Figure 4 Evolution and performance of nucleocapsids with exterior surface mutations in vitro or in vivo.
a, Heat map of log enrichments between the injected pool and RNA recovered from the tail vein 60 min later. Purple and orange indicate mutations that were depleted or enriched in the selected population, respectively. Blue squares and black dots indicate the I53-50-v3 starting sequence and I53-50-v4 selected sequence, respectively. Residues not in the designed combinatorial library are coloured grey. Note the strong enrichment of the E67K mutation and corresponding depletion of the native E67 allele. b, Design model of I53-50-v4. Colouring is as described in Fig. 1a. c, Four variants were tested: a consensus sequence based on the most common residue at each position after selection in mouse circulation (consensus, I53-50-v4), the full-length sequence with the greatest fold increase in population fraction (Most_enriched), the sequence with the most total counts (Top_count), and I53-50-v3 with only the E67K mutation (v3_E67K). Previous versions (I53-50-v1, I53-50-v2 and I53-50-v3) were also included as benchmarks. Each variant was individually expressed and purified by IMAC before being pooled (equal protein concentration) and purified en masse by SEC. The resulting nucleocapsid pool was then incubated in whole blood (n = 3 independent reactions per time point). RNA was recovered at the indicated time points, and the fraction of each variant was determined by Illumina MiSeq counts taken at each time point. d, The same nucleocapsid pool used in c was injected retro-orbitally into mice (n = 5 biologically independent mice). I53-50-v3 was evaluated with (v3) and without (v3H) the H6Q and H9Q mutations, and both variants were found to have similar behaviour. Error bars represent s.e.m.
Extended Data Figure 5 Negative-stain transmission electron microscopy class averages.
a, Two-dimensional class averages of I53-50-v0 (7,979 particles) and I53-50-v4 (7,120 particles) data sets showing the percentage of the total particles present in each class. I53-50-v4 nucleocapsids are on average denser than unfilled I53-50-v0 assemblies, especially near the inner surface of the capsid. b, All I53-50-v0 and I53-50-v4 particles from a were combined into a single set (15,119 particles), and 20 class averages were made from the combined data. Class averages were grouped into three bins (v0 dominant has ≤ 25% I53-50-v4, v4 dominant has ≥ 74% I53-50-v4, and mixed has the rest) and arranged from left to right with increasing fraction of I53-50-v4 particles (shown below each class). The v0 dominant classes appear more similar to the I53-50-v0 class averages in a, while the v4 dominant classes appear more similar to the I53-50-v4 class averages. The percentage of the complete I53-50-v4 dataset found in each class is shown above each class average. c, Table presenting the bins into which I53-50-v4 particles were assigned. We found that 64% of I53-50-v4 particles were present in the v4 dominant classes, which also seem to be more filled than the v0-dominant classes. Although TEM cannot determine the nature of the contents, encapsulated RNA is plausible.
Extended Data Figure 6 Summary of encapsulated RNA composition analysis.
a, Flow chart explaining the relationship between bulk RNA measurements and RT–qPCR quantification. Bulk RNA measurements also account for cellular RNA and nucleocapsid genome fragments, whereas RT–qPCR only quantifies full-length genomes. Ratios of nucleocapsid genomes to capsids are based on these measurements and are reported in parentheses. b, Stacked bar plot describing the fractions of total encapsulated RNA that are full-length nucleocapsid genomes or fragments thereof.
Extended Data Figure 7 Packaging correlates strongly with expression level in producer cells.
a, log enrichment (fraction packaged in nucleocapsid divided by fraction produced in cells) for I53-50-v4 versus I53-50-v1. Each point represents a unique RNA (red squares are protein coding mRNAs, green triangles are non-coding RNAs such as ribosomal RNA, and the blue circle is the nucleocapsid genomic RNA). No increase in specificity was observed over the course of evolution from the rationally designed I53-50-v1 to the in vivo circulating I53-50-v4. This is not surprising because no attempt was made to evolve increased specificity. The diagonal line is y = x. b, log fraction of total reads in nucleocapsids versus log fraction of total reads in cells shows that packaging correlates strongly with expression level (Pearson values for I53-50-v1 and I53-50-v4 are 0.83 and 0.86, respectively). Each point represents a unique RNA. The diagonal line is y = x. RNAs above the line are enriched in nucleocapsids, and RNAs below the line are depleted in nucleocapsids. Although the nucleocapsid genome is slightly enriched, its high packaging yield seems to arise because T7 RNA polymerase floods the cell with genomes, thereby increasing the chance that the capsid randomly packages the genome. Conversely, ribosomal RNA may be restricted from nucleocapsids because intact ribosomes are too large to be encapsulated. All data points represent the average of two independent biological replicates.
Extended Data Figure 8 Design models of synthetic nucleocapsid versions 1 to 4.
Trimer subunits are coloured green and pentamer subunits are coloured cyan. Mutations with respect to the previous version are coloured blue (increases in positive charge and/or decreases in negative charge (for example, E→N, N→K and E→K)), orange (no change in charge (for example, E→D, N→T and K→R)), or red (decreases in positive charge and/or increases in negative charge (for example, N→E, K→N, K→E)).
Extended Data Figure 9 Dynamic light scattering of nucleocapsids.
Dynamic light scattering (DLS) was performed on synthetic nucleocapsids and fitted with regularization analysis, confirming uniform populations of nucleocapsids around the expected size. a, I53-50-v0 has a C-terminal histidine tag. b, I53-50-v1 has an N-terminal histidine tag that was cleaved before DLS. c, I53-50-v4 has an N-terminal histidine tag that was cleaved before DLS. The experiment was independently repeated three times (data for independent replicates are shown in the figure).
Supplementary information
Supplementary Information
This file contains a list of supplemental figures S1-S10, and supplemental tables S1-S6. (PDF 26998 kb)
Supplementary Table 1
This file contains supplementary table 1 - composition of libraries produced for each step of evolution. (XLSX 14 kb)
Supplementary Table 2
This file contains supplementary table 2 - protein sequences of hydrophilic peptide library for increasing circulation time in live mice. (XLSX 41 kb)
Rights and permissions
About this article

Cite this article
Butterfield, G., Lajoie, M., Gustafson, H. et al. Evolution of a designed protein assembly encapsulating its own RNA genome. Nature 552, 415–420 (2017). https://doi.org/10.1038/nature25157
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature25157
This article is cited by
-
Protein-modified nanomaterials: emerging trends in skin wound healing
Discover Nano (2023)
-
Drug delivery systems for CRISPR-based genome editors
Nature Reviews Drug Discovery (2023)
-
De novo design of protein structure and function with RFdiffusion
Nature (2023)
-
In silico evolution of nucleic acid-binding proteins from a nonfunctional scaffold
Nature Chemical Biology (2022)
-
Genetically engineered materials: Proteins and beyond
Science China Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
