
Abstract
Our growing awareness of the microbial world’s importance and diversity contrasts starkly with our limited understanding of its fundamental structure. Despite recent advances in DNA sequencing, a lack of standardized protocols and common analytical frameworks impedes comparisons among studies, hindering the development of global inferences about microbial life on Earth. Here we present a meta-analysis of microbial community samples collected by hundreds of researchers for the Earth Microbiome Project. Coordinated protocols and new analytical methods, particularly the use of exact sequences instead of clustered operational taxonomic units, enable bacterial and archaeal ribosomal RNA gene sequences to be followed across multiple studies and allow us to explore patterns of diversity at an unprecedented scale. The result is both a reference database giving global context to DNA sequence data and a framework for incorporating data from future studies, fostering increasingly complete characterization of Earth’s microbial diversity.
Similar content being viewed by others


Main
A primary aim of microbial ecology is to determine patterns and drivers of community distribution, interaction, and assembly amidst complexity and uncertainty. Microbial community composition has been shown to change across gradients of environment, geographic distance, salinity, temperature, oxygen, nutrients, pH, day length, and biotic factors1,2,3,4,5,6. These patterns have been identified mostly by focusing on one sample type and region at a time, with insights extrapolated across environments and geography to produce generalized principles. To assess how microbes are distributed across environments globally—or whether microbial community dynamics follow fundamental ecological ‘laws’ at a planetary scale—requires either a massive monolithic cross-environment survey or a practical methodology for coordinating many independent surveys. New studies of microbial environments are rapidly accumulating; however, our ability to extract meaningful information from across datasets is outstripped by the rate of data generation. Previous meta-analyses have suggested robust general trends in community composition, including the importance of salinity1 and animal association2. These findings, although derived from relatively small and uncontrolled sample sets, support the utility of meta-analysis to reveal basic patterns of microbial diversity and suggest that a scalable and accessible analytical framework is needed.
The Earth Microbiome Project (EMP, http://www.earthmicrobiome.org) was founded in 2010 to sample the Earth’s microbial communities at an unprecedented scale in order to advance our understanding of the organizing biogeographic principles that govern microbial community structure7,8. We recognized that open and collaborative science, including scientific crowdsourcing and standardized methods8, would help to reduce technical variation among individual studies, which can overwhelm biological variation and make general trends difficult to detect9. Comprising around 100 studies, over half of which have yielded peer-reviewed publications (Supplementary Table 1), the EMP has now dwarfed by 100-fold the sampling and sequencing depth of earlier meta-analysis efforts1,2; concurrently, powerful analysis tools have been developed, opening a new and larger window into the distribution of microbial diversity on Earth. In establishing a scalable framework to catalogue microbiota globally, we provide both a resource for the exploration of myriad questions and a starting point for the guided acquisition of new data to answer them. As an example of using this tool, we present a meta-analysis of the EMP archive, tracking individual sequences across diverse samples and studies with standardized environmental descriptors, investigating large-scale ecological patterns, and exploring key hypotheses in ecological theory to serve as seeds for future research.
A standardized and scalable approach
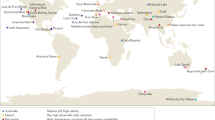
The EMP solicited the global scientific community for environmental samples and associated metadata spanning diverse environments and capturing spatial, temporal, and/or physicochemical covariation. The first 27,751 samples from 97 independent studies (Supplementary Table 1) represent diverse environment types (Fig. 1a), geographies (Fig. 1b), and chemistries (Extended Data Fig. 1). The EMP encompasses studies of bacterial, archaeal, and eukaryotic microbial diversity. The analysis here focuses exclusively on the bacterial and archaeal components of the overall database (for concision, use of ‘microbial’ will hereafter refer to bacteria and archaea only). Associated metadata included environment type, location information, host taxonomy (if relevant), and physicochemical measurements (Supplementary Table 2). Physicochemical measurements were made in situ at the time of sampling. Investigators were encouraged to measure temperature and pH at minimum. Salinity, oxygen, and inorganic nutrients were measured when possible, and investigators collected additional metadata pertinent to their particular investigations.

a, The EMP ontology (EMPO) classifies microbial environments (level 3) as free-living or host-associated (level 1) and saline or non-saline (if free-living) or animal or plant (if host-associated) (level 2). The number out of 23,828 samples in the QC-filtered subset in each environment is provided. EMPO is described with examples at http://www.earthmicrobiome.org/protocols-and-standards/empo. b, Global scope of sample provenance: samples come from 7 continents, 43 countries, 21 biomes (ENVO), 92 environmental features (ENVO), and 17 environments (EMPO).
Metadata were required to conform to the Genomic Standards Consortium’s MIxS and Environment Ontology (ENVO) standards10,11. We also used a light-weight application ontology built on top of ENVO: the EMP Ontology (EMPO) of microbial environments. EMPO was tailored to capture two major environmental axes along which microbial beta-diversity has been shown to orient: host association and salinity1,2. We indexed the classes in this application ontology (Fig. 1a) as levels of a structured categorical variable to classify EMP samples as host-associated or free-living (level 1). Samples were categorized within those classes as animal-associated versus plant-associated or saline versus non-saline, respectively (level 2). A finer level (level 3) was then assigned that satisfied the degree of environment granularity sought for this meta-analysis (for example, sediment (saline), plant rhizosphere, or animal distal gut). We expect EMPO to evolve as new studies and sample types are added to the EMP and as additional patterns of beta-diversity are revealed.
We surveyed bacterial and archaeal diversity using amplicon sequencing of the 16S rRNA gene, a common taxonomic marker for bacteria and archaea12 that remains a valuable tool for microbial ecology despite the introduction of whole-genome methods (for example, shotgun metagenomics) that capture gene-level functional diversity13. DNA was extracted from samples using the MO BIO PowerSoil DNA extraction kit, PCR-amplified, and sequenced on the Illumina platform. Standardized DNA extraction was chosen to minimize the potential bias introduced by different extraction methodologies; however, extraction efficiency may also be subject to interactions between sample type and cell type, and thus extraction effects should be considered as a possible confounding factor in interpreting results. We amplified the 16S rRNA gene (V4 region) using primers14 shown to recover sequences from most bacterial taxa and many archaea15. We note that these primers may miss newly discovered phyla with alternative ribosomal gene structures16, and subsequent modifications not used here have shown improved efficiency with certain clades of Alphaproteobacteria and Archaea17,18,19. We generated sequence reads of 90–151 base pairs (bp) (Extended Data Fig. 2a, Supplementary Table 1), totaling 2.2 billion sequences, an average of 80,000 sequences per sample.
Sequence analysis and taxonomic profiling were done initially using the common approach of assigning sequences to operational taxonomic units (OTUs) clustered by sequence similarity to existing rRNA databases14,20. While this approach was useful for certain analyses, for many sample types, especially plant-associated and free-living communities, one-third of reads or more could not be mapped to existing rRNA databases (Extended Data Fig. 2b). We therefore used a reference-free method, Deblur21, to remove suspected error sequences and provide single-nucleotide resolution ‘sub-OTUs’, also known as ‘amplicon sequence variants’22, here called ‘tag sequences’ or simply ‘sequences’. Because Deblur tag sequences for a given meta-analysis must be the same length in each sample, and some of the EMP studies have read lengths of 90 bp, we trimmed all sequences to 90 bp for this meta-analysis. We verified that the patterns presented here were not adversely affected by trimming the sequences (Extended Data Fig. 3). As we show, 90-bp sequences were sufficiently long to reveal detailed patterns of community structure. Because exact sequences are stable identifiers, unlike OTUs, they can be compared to any 16S rRNA or genomic database now and in the future, thereby promoting reusability22.
Microbial ecology without OTU clustering
While earlier large-scale 16S rRNA amplicon studies adopted OTU clustering approaches in part out of concern that erroneous reads would dominate diversity assessments23, patterns of prevalence (presence–absence) in our results suggest that Deblur error removal produced ecologically informative sequences without clustering. After rarefying to 5,000 sequences per sample, a total of 307,572 unique sequences were contained in the 96 studies and 23,828 samples of the ‘QC-filtered’ Deblur 90-bp observation table. Among studies, more than half (57%) of all obtained sequences were observed in two or more studies, but only 5% were observed in more than ten studies; the most prevalent sequence was found in 88 of 96 studies (Extended Data Fig. 4a). Among samples, although most sequences (86%) were observed in two or more samples, only 7% were observed in more than 100 samples (Extended Data Fig. 4b). As expected, the most prevalent sequences were also the most abundant (Extended Data Fig. 4c).
Our analyses were carried out using a modest sequencing depth of 5,000 observations per sample after Deblur and rarefaction. To investigate how prevalence estimates were affected by sequencing depth, we focused on four major environment types for which we had the greatest number of samples with more than 50,000 observations (soil, saltwater, freshwater, and animal distal gut). The relationship between average tag sequence prevalence and sequencing depth differed among these environments (Extended Data Fig. 4d) but was generally positive, suggesting that our global analysis underestimated true prevalence. Animal-associated microbiomes were a notable exception, with an upper bound on prevalence apparently imposed by host specificity when all host species were considered (Extended Data Fig. 4e); this bound disappeared when considering only human-derived samples (Extended Data Fig. 4f). Although contamination remains an issue in microbiome studies24, most of the very highly abundant and prevalent sequences here had higher mean relative abundances among samples than among no-template controls (Supplementary Table 3), suggesting that they did not originate from reagents.
Matches between our sequences and existing 16S rRNA gene reference databases highlight the novelty captured by the EMP. Exact matches to 46% of Greengenes25 and 45% of SILVA26 rRNA gene databases were found in our dataset, indicating that we ‘recaptured’ nearly half of the reference sequence diversity with just under 100 environmental surveys. These matches accounted for 10% and 13%, respectively, of the tag sequences in our dataset, indicating that large swathes of microbial community diversity are not yet captured in full-length sequence databases. The failure of many sequences to be mapped in reference-based alignments to Greengenes and SILVA 97% identity OTUs (Extended Data Fig. 2b) supports this observation.
Patterns of diversity reflect environment
We used a structured categorical variable of microbial environments, EMPO, to analyse diversity in the EMP catalogue in the context of lessons from previous investigations1,2. We observed environment-dependent patterns in the number of observed tag sequences (alpha-diversity), turnover and nestedness of taxa (beta-diversity), and predicted genome properties (ecological strategy). Derived from a more standardized methodology, our dataset confirms the previous finding2 that host association is a fundamental environmental factor that differentiates microbial communities (Fig. 2c, Extended Data Fig. 2d). We build on this pattern by showing that there is less richness in host-associated communities than in free-living communities (Fig. 2a), with the noted exception of plant rhizosphere samples, which resemble free-living soil communities in both richness (Fig. 2a) and composition (Fig. 2c). Our findings also confirm the major compositional distinction between saline and non-saline communities1 (Fig. 2c). The effect sizes of environmental factors on alpha- and beta-diversity generally showed large contributions of environment type and (for host-associated samples) host species to both types of diversity (Extended Data Fig. 5a, b).

a, Within-community (alpha) diversity, measured as number of observed 90-bp tag sequences (richness), in n = 23,828 biologically independent samples as a function of environment (per-environment n shown in Fig. 1a), with boxplots showing median, interquartile range (IQR), and 1.5 × IQR (with outliers). Tag sequence counts were subsampled to 5,000 observations. Yellow line indicates the median number of observed tag sequences for all samples in that set of boxplots. Free-living communities of most types exhibited greater richness than host-associated communities. b, Tag sequence richness (as in a) versus pH and temperature in n = 3,986 (pH) and n = 6,976 (temperature) biologically independent samples. Black points are the 99th percentiles for richness across binned values of pH and temperature. Laplace (two-sided exponential) curves captured apparent upper bounds on microbial richness and their peaked distributions better than Gaussian curves. Greatest maximal richness occurred at values of pH and temperature that corresponded to modes of the Laplace curves. Maximum richness exponentially decreased away from these apparent optima. c, Between-community (beta) diversity among in n = 23,828 biologically independent samples: principal coordinates analysis (PCoA) of unweighted UniFrac distance, PC1 versus PC2 and PC1 versus PC3, coloured by EMPO levels 2 and 3. Clustering of samples could be explained largely by environment. d, 16S rRNA gene average copy number (ACN, abundance-weighted) of EMP communities in n = 23,228 biologically independent samples, coloured by environment. EMPO level 2 (left): animal-associated communities had a higher ACN distribution than plant-associated and free-living (both saline and non-saline) communities. Right: soil communities had the lowest ACN distribution, while animal gut and saliva communities had the highest ACN distribution.
The ability to identify sample provenance using only a microbial community profile has applications ranging from criminal forensics to mistaken sample identification; these applications will require large curated datasets, such as the EMP. Supervised machine learning demonstrated that samples could be distinguished as being animal-associated, plant-associated, saline free-living, or non-saline free-living with 91% accuracy based solely on community composition, and to fine-scale environment with 84% accuracy (Extended Data Fig. 5c–e). The most commonly misclassified samples were soil, non-saline surface and aerosol, and animal secretion. In many of these cases, misclassification can be attributed to the limitations of EMPO. As additional samples are classified, classification can be improved by iteratively and empirically redefining categories using machine learning. Conversely, with continuous factors, such as salinity, categorical definitions cannot perfectly capture intermediate values. High classification success to environment type was supported by source-tracking analyses (Extended Data Fig. 5f, g), with the exception of plant rhizosphere samples, owing to their similarity to soil samples.
Predicted average community copy number (ACN) of the 16S rRNA gene was another metric found to differentiate microbial communities in both host-associated and free-living communities (Fig. 2d). ACN can be predicted from 16S rRNA amplicon data27; this method has been used, for example, to link the taxonomic groups associated with copiotrophic and oligotrophic behaviours in soils to high and low rRNA gene copy numbers, respectively28. Approximately half the dataset centred on an ACN of 2.2 (free-living and plant-associated samples) and the other half on an ACN of 3.4 (animal-associated samples) (Fig. 2d). Greater per-genome rRNA operon copy number has been found to be associated with rapid maximum growth rates29, which may provide a selective advantage when resources are abundant, such as in animal hosts. While ACN is an estimate rather than a measurement of average rRNA copy number and is subject to potential biases in the underlying reference database, the distributions we observed are consistent with 16S rRNA copy number reflecting differences in ecological strategies among environments.
A resource for theoretical ecology
The coordinated accumulation of data across studies allows investigations of patterns within (alpha-diversity) and among (beta-diversity) microbial communities at scales that vastly exceed what could be measured in any individual study. Patterns of alpha-diversity in meta-analyses have revealed global trends that have been key to the development of major ideas in macroecological theory, but fundamental patterns have been more difficult to discern in microbial ecology. For example, a nearly ubiquitous tendency towards greater diversity in the tropics is evident in macroecology30, but there is substantial variation among studies examining latitudinal trends of microbial diversity31,32,33. The large EMP dataset analysed here reveals a weak but significant trend towards increasing diversity at lower latitudes in non-host-associated environments (Extended Data Fig. 5h). An effect of latitude was apparent both within and across studies, consistent with global trends in latitudinal microbial diversity being an emergent function of locally selective environmental heterogeneity34. However, substantial study-to-study variation in richness highlights the caveats inherent in meta-analysis; more coordination of sample collections from similar environments across larger gradients is necessary to better address this question.
The EMP has the potential to link global patterns of microbial diversity with physicochemical parameters—if appropriate metadata are provided by researchers. Microbial community richness has been found to correlate with environmental factors, including pH and temperature3,33,35,36. For example, richness has been shown to increase up to neutral pH36 and often to decrease above neutral pH3,35 in soil communities. Richness has been shown to increase with temperature up to a limit and then to decrease beyond that limit in seawater (maximum at about 19 °C)33 and to increase with temperature in soil (up to at least around 26 °C)36. However, general relationships of richness to temperature and pH remain unresolved37. Here, across samples from non-host-associated environments where pH or temperature were measured (mostly freshwater and soil environments), richness was greatest near neutral pH (around 7) and relatively cool temperatures (about 10 °C) (Fig. 2b). We observed apparent upper bounds on richness with both temperature and pH that were best fit by two-sided exponential (Laplace) curves. Thus, the present dataset suggests that maximum microbial richness occurs within a relatively narrow range of intermediate pH and temperature values. These patterns, while robust in the context of the EMP dataset, necessarily reflect only the subset of sample types for which variables were measured (Supplementary Table 2); they should therefore be interpreted with caution. Understanding universal relationships between richness and environmental factors will require information from more studies with detailed and carefully collected physicochemical metadata.
Beyond measured physical covariates, the breadth of environments in the EMP catalogue allows a detailed exploration of how microbial diversity is distributed across environments. Diversity among communities (beta-diversity) is driven by turnover (replacement of taxa) and nestedness (gain or loss of taxa resulting in differences in richness)38. If turnover dominates, then disparate communities will harbour unique taxa. If nestedness dominates, then communities with fewer taxa will be subsets of communities with more taxa. We tested for nestedness using a 2,000-sample subset with even representation across environments and studies. Given the contrasting environments and geographic separation among the many studies in the EMP, we expected different environments to contain unique sets of taxa and to show little nestedness. However, we found that communities across environments were significantly nested (Fig. 3a, b; P < 0.05) in comparison to null models (Fig. 3c), accounting for the observed patterns of richness. At coarse taxonomic levels, an average of 84% of phyla, 73% of classes, and 58% of orders that occurred in less diverse samples also occurred in more diverse samples. Nestedness was observed even when the most prevalent taxa were removed and was robust across randomly chosen subsets of samples (Extended Data Fig. 6). These patterns could have resulted from several mechanisms, including ordered extinctions39 and the filtering of complex communities over time40, differential dispersal abilities41 and cascading source–sink colonization processes that assemble nested subsets from more complex communities, or by the tendency of larger habitat patches to support more rare taxa with lower prevalence42. Notably, finer taxonomic groupings showed less nestedness (Fig. 3c), indicating that the processes that underlie nested patterns of turnover are likely to reflect conserved aspects of microbial biology, and not to result from the interplay of diversification and dispersal on short timescales.

a, Presence–absence of phyla across samples, with phyla (rows) sorted by prevalence and samples (columns) sorted by richness. Shown is a subset of the EMP consisting of n = 2,000 biologically independent samples with even representation across environments and studies. With increasing sample richness (left to right), phyla tended to be gained but not lost (P < 0.0001 versus null model; NODF (nestedness measure based on overlap and decreasing fills) statistic and 95% confidence interval = 0.841 ± 0.018). b, As in a but separated into non-saline, saline, animal, and plant environments (P < 0.0001, respective NODF = 0.811 ± 0.013, 0.787 ± 0.015, 0.788 ± 0.018 and 0.860 ± 0.021). c, Nestedness as a function of taxonomic level, from phylum to tag sequence, across all samples and within environment types. Also shown are median null model NODF scores (± s.d.). NODF measures the average fraction of taxa from less diverse communities that occur in more diverse communities. All environments at all taxonomic levels were more nested than expected randomly, with nestedness higher at higher taxonomic levels (for example, phyla).
These global ecological patterns offer a glimpse of what is possible with coordinated and cumulative sampling—in addition to the specific questions addressed by individual studies, context is built and easily queried across studies. They also necessarily highlight the inherent limitations to decentralized studies, especially regarding the collection of comparable environmental data. Future studies will be able to use the current EMP data as a starting point for more explicit tests of broad ecological principles, both to identify gaps in current knowledge and to more confidently plan large directed studies with sufficient power to fill them.
A more precise and scalable catalogue
An advantage of using exact sequences is that they enable us to observe and analyse microbial distribution patterns at finer resolution than is possible with traditional OTUs. As an example, we applied a Shannon entropy analysis to tag sequences and higher taxonomic groups to measure biases in the distribution of taxa. Taxa that are equally likely to be found in any environment will have high entropy and low specificity, whereas taxa found only in a single environment will have low entropy and high specificity (note that we use ‘specificity’ solely to denote distributional patterns, not to imply adaptation or causality). Tag sequences exhibited high specificity for environment, with distributions skewed towards one or a few environments (low Shannon entropy); by contrast, higher taxonomic levels tended to be more evenly distributed across environments (high Shannon entropy, low specificity) (Fig. 4a). Entropy distributions across all tag sequences at each taxonomic level show that this pattern is general (Fig. 4b). Seeking a more precise measure of the divergence at which a taxon is specific for environments, we next investigated how entropy changes as a function of phylogenetic distance. We calculated entropy for each node of the phylogeny and visualized it as a function of maximum tip-to-tip branch length (Fig. 4c). While entropy decreased gradually at finer phylogenetic resolution, it dropped sharply at the tips of the tree. We conclude that environment specificity is best captured by individual 16S rRNA sequences, below the typical threshold defining microbial species (97% identity of the 16S rRNA gene).

a, Environment distribution in all genera and 400 randomly chosen tag sequences, drawn from n = 2,000 biologically independent samples with even representation across environments (EMPO level 3) and studies. Each bar depicts the distribution of environments for a taxon (not relative abundance of taxa): bars composed mostly of one colour (environment) are specific for that environment, as seen with tag sequences; bars composed of many colours are more cosmopolitan, as seen with genera. Tag sequences were more specific for environment than were genera and higher taxonomic levels. b, Shannon entropy within each taxonomic group (minimum 20 tag sequences per group) and for the same set of samples with permuted taxonomy labels. Box plots show median, IQR, and 1.5 × IQR (with outliers) for each taxonomic level. A violin plot shows the entropy of tag sequences (minimum 10 samples per tag sequence). Specificity for environment occurred predominantly below the genus level. c, Shannon entropy within phylogenetic subtrees of tag sequences (minimum 20 tips per subtree) defined by maximal tip-to-tip branch length (substitutions per site) and for the same samples with permuted phylogenetic tree tips. Mean and 20th/80th percentile for a sliding window average of branch length is shown. Violin plot for tag sequences as in b. Dotted lines show average tip-to-tip branch length corresponding to 97% sequence identity and taxonomic levels displayed in b. The greatest decrease in entropy was between the lowest branch length subtree tested and tag sequences.
The EMP dataset provides the ability to track individual sequences across the Earth’s microbial communities. Using a representative subset of the EMP (Extended Data Fig. 7a), we produced a table of sequence counts and distributions, including among environments (EMPO) and along environmental gradients (pH, temperature, salinity, and oxygen). From this we generated ‘EMP Trading Cards’, which promote exploration of the dataset and here highlight the distribution patterns of three prevalent or environment-correlated tag sequences (Extended Data Fig. 7b, Supplementary Table 3). The entire EMP catalogue can be queried using the Redbiom software, with command-line (https://github.com/biocore/redbiom) and web-based (http://qiita.microbio.me) interfaces to find samples based on sequences, taxa, or sample metadata, and to export selected sample data and metadata (instructions at https://github.com/biocore/emp). User data generated from the EMP protocols can be readily incorporated into this framework: because Deblur operates independently on each sample21, additional tag sequences can be added to this dataset from new studies without reprocessing existing samples. Future combinations of datasets targeting the same genomic region but sequenced using different methods may be admissible but would require considerations to account for methodological biases.
The growing EMP catalogue is expected to have applications for research and industry, with tag sequences used as environmental indicators and representing targets for cultivation, genome sequencing, and laboratory study. In addition, these tools and approaches, although developed for bacteria and archaea, could be expanded to all domains of life43. To achieve greater utility for the EMP and similar projects, we must continually improve metadata collection and curation, ontologies, support for multi-omics data, and access to computational resources.
Conclusions and future directions
Here we have used crowdsourced sample collection and standardized microbiome sequencing and metadata curation to perform a global meta-analysis of bacterial and archaeal communities. Using exact sequences in place of OTUs and a learned structure of microbial environments, we have shown that agglomerative sampling can reveal basic biogeographic patterns of microbial ecology, with resolution and scope rivaling data compilations currently available for ‘macrobial’ ecology44,45. Our results point to key organizing principles of microbial communities, with less-rich communities nested within richer communities at higher taxonomic levels, and environment specificity becoming much more evident at the level of individual 16S rRNA sequences.
The EMP framework and global synthesis presented here represent value added to the scientific community beyond the substantial contributions of the constituent studies (Supplementary Table 1). However, as with any meta-analysis in which data are gathered primarily in service of separate questions rather than a single theme46, conclusions should be viewed with caution and form starting points for future hypothesis-directed investigations. There is a need to span gradients of geography (for example, latitude and elevation) and chemistry (for example, temperature, pH, and salinity) more evenly—assisted by tools for more comprehensive collection and curation of metadata—and to track environments over time. In addition, biotic factors (for example, animals, fungi, plants, viruses, and eukaryotic microbes) not measured in this study have important roles in determining community structure4,5,6. The scalable framework introduced here can be expanded to address these needs: new studies to fill gaps in physicochemical space, amplicon data for microbial eukaryotes and viruses, and whole-genome and whole-metabolome profiling. At a time when both academic and governmental agencies increasingly recognize the value of communal biodiversity monitoring efforts47,48, the EMP provides one example of a logistically feasible standardization framework to maximize interoperability across diverse and independent studies, in particular using stable identifiers (exact sequences) to enable enduring utility of environmental sequence data. Given current global sequencing efforts, the use of coordinated protocols and submission to this and other public databases should allow rapid accumulation of new data, providing an ever more diverse reference catalogue of microbes and microbiomes on Earth.
Methods
Study design
This effort was possible because of a unified standard workflow that leveraged existing sample and data reporting standards to allow biomass and metadata collection across diverse environments on Earth. After sample collection, all samples were processed following the same protocols. A standard DNA extraction protocol (http://www.earthmicrobiome.org/protocols-and-standards/dna-extraction-protocol) was implemented to ensure that trends observed were due either to the biological system or to biases in extraction potential for organisms from different environmental matrices, and not due to inherent biases in the extraction protocol. To avoid known issues that arise when multiple amplicon strategies are combined49, we also standardized PCR primers, amplification strategy, and sequencing50. More recent studies not included in this meta-analysis adopted additional primer modifications to allow recovery of key taxa in marine and soil samples17,18,19. Data reporting standards, including the MIxS (minimal information about any sequence) metadata standard developed by the Genomic Standards Consortium10 and the Environment Ontology (ENVO)11,51, enabled interoperability, data analysis, and interpretation between samples from disparate environments, collected using many different techniques through unconnected programs of investigation.
To transfer our knowledge of microbial environments to the broader community, we engaged with the developers of ENVO to ensure that the basic, salient features of microbial environments (host-associated or free-living, and respectively within those, animal- or plant-associated, and saline or non-saline materials) were represented either in this ontology or in those with which it interoperates. For ease of application, we gathered these contributions into an application ontology, the EMP Ontology (EMPO) (Fig. 1a). The EMP community will continue to work with ontology engineers to shape ENVO and other ontologies around the EMPO application ontology. EMPO will be maintained as a logical subset of ENVO and integrated into the ENVO release cycle to maximize interoperation.
Metadata curation was automated using Pandas (http://pandas.pydata.org). The size of the dataset also required extensive software development to support analysis at this scale, leading to tools including the data and analysis portal Qiita (http://qiita.microbio.me), the BIOM format52, new ‘OTU picking’ methods Deblur21 and a subsampled open-reference procedure53, a scalability improvement of Fast UniFrac phylogenetic inference software54, speed improvements to sequence-insertion tree method SEPP55, and speed and feature improvements to Emperor ordination visualization software56 (http://biocore.github.io/emperor).
Sample collection
The global community of microbial ecologists was invited to submit samples for microbiome analysis, and samples were accepted for DNA extraction and sequencing provided that scientific justification and high-quality sample metadata were provided before sample submission. Standardized sampling procedures for each sample type were used by contributing investigators. Samples were collected fresh and, where possible, immediately frozen in liquid nitrogen and stored at –80 °C. Detailed sampling protocols are described in publications of the individual studies (Supplementary Table 1). Bulk samples (for example, soil, sediment, faeces) and fractionated bulk samples (for example, sponge coral surface tissue, centrifuged turbid water) were taken using microcentrifuge tubes. Swabs (BD SWUBE dual cotton swabs or similar) were used for biofilm or surface samples. Filters (Sterivex cartridges, 0.2 μm, Millipore) were used for water samples. Samples were sent to laboratories in the United States for DNA extraction and sequencing: water samples to Argonne National Laboratory, soil samples to Lawrence Berkeley National Laboratory (pre-2014) or Pacific Northwest National Laboratory (2014 onward), and faecal and other samples to the University of Colorado Boulder (pre-2015) or the University of California San Diego (2015 onward).
Metadata curation and EMP ontology
Metadata were collected in compliance with MIMARKS10, EBI (https://www.ebi.ac.uk/ena), and Qiita (http://qiita.microbio.me) standards, as described in the EMP Metadata Guide (http://www.earthmicrobiome.org/protocols-and-standards/metadata-guide). QIIME mapping files (metadata) were downloaded from Qiita, merged, and refined using Python with Pandas, generating quality-controlled mapping files. Mapping file columns are described in Supplementary Table 2. Mapping files for the full EMP dataset and subsets (see below) are available at ftp://ftp.microbio.me/emp/release1/mapping_files/. The EMP Ontology (EMPO) for microbial environments was devised to facilitate the present analysis while preserving interoperability. Coordinated by the ENVO team, annotations from ENVO11,51, UBERON (metazoan anatomy)57, PO (plant ontology)58, FAO (fungal anatomy ontology, http://purl.obolibrary.org/obo/fao.owl), and OMP (ontology of microbial phenotypes)59 were mapped to our EMPO levels 2 and 3 (empo_2 and empo_3). Additionally, the free-living or host-associated lifestyles were captured in level 1 categories (empo_1). Descriptions of empo_3 categories are provided at http://www.earthmicrobiome.org/protocols-and-standards/empo. The W3C Web Ontology Language (OWL) document is available at http://purl.obolibrary.org/obo/envo/subsets/envoEmpo.owl. Map data were derived from the open-source project MatPlotLib package Basemap, which distributes map data from Generic Mapping Tools data (http://gmt.soest.hawaii.edu) released under the GNU Lesser General Public License v3.
DNA extraction, amplicon PCR, sequencing, and sequence pre-processing
DNA extraction and 16S rRNA amplicon sequencing was done using EMP standard protocols (http://www.earthmicrobiome.org/protocols-and-standards/16s)14. In brief, DNA was extracted using the MO BIO PowerSoil DNA extraction kit (Carlsbad, CA), chosen because of its versatility with diverse sample types (rather than high yields with any given sample type). Amplicon PCR was performed on the V4 region of the 16S rRNA gene using the primer pair 515f–806r50 with Golay error-correcting barcodes on the reverse primer. Although any primer-based method necessarily under-samples diversity, a recent analysis of 16S rRNA genes captured in shotgun metagenomic sequences indicates that this primer pair is among the best available for sampling both bacteria and non-eukaryotic archaea15. Amplicons were barcoded and pooled in equal concentrations for sequencing. The amplicon pool was purified with the MoBio UltraClean PCR Clean-up kit and sequenced on the Illumina HiSeq or MiSeq sequencing platform; the same sequencing primers were used with both platforms, and previous work has shown that conclusions drawn from 16S rRNA amplicon data are not dependent on which of these sequencing platforms is used50. Sequence data were demultiplexed and minimally quality filtered using the QIIME 1.9.1 script split_libraries_fastq.py60 with Phred quality threshold of 3 and default parameters to generate per-study FASTA sequence files.
Tag sequence and OTU picking and subsets
Sequence data were error-filtered and trimmed to the length of the shortest sequencing run (90 bp) using the Deblur software21; the resulting 90-bp Deblur BIOM table was used for all analyses unless otherwise noted. Deblur tables trimmed to 100 bp and 150 bp were also generated and provided, which contain greater sequence resolution but fewer samples. Deblur observation tables were filtered to keep only tag sequences with at least 25 reads total over all samples. For comparison to existing OTU tables, traditional closed-reference OTU picking was done against 16S rRNA databases Greengenes 13.825 and SILVA 12326 using SortMeRNA61, and subsampled open-reference OTU picking53 was done against Greengenes 13.8. These unfiltered tables and the filtered and subset tables described below are available at ftp://ftp.microbio.me/emp/release1/otu_tables.
A total of 97 studies and 27,742 samples are included in the present study and in the unfiltered BIOM tables. The QC-filtered subset used in core diversity analyses (Fig. 2) contains 96 studies and 23,828 samples, and it was subset further for some analyses. In the provided BIOM tables (ftp://ftp.microbio.me/emp/release1/otu_tables/ and https://zenodo.org/record/890000), the ‘release1’ set contains all samples in the 97 studies that have at least one sequence per sample; this set includes controls (blanks and mock communities). The ‘qc_filtered’ set, from which the subsets are drawn, has samples with ≥ 1,000 observations in each of four observation tables: closed-reference Greengenes, closed-reference SILVA, open-reference Greengenes, and Deblur 90-bp; controls (empo_1 == ‘Control’) are excluded. Subsets were then generated which give equal (as possible) representation across environments (EMPO level 3) and across studies within those environments. The subsets contain 10,000, 5,000, and 2,000 samples (nested subsets). In each subset the samples must have ≥ 5,000 observations in the Deblur 90-bp observation table and ≥ 10,000 observations in each of the closed-reference Greengenes, closed-reference SILVA, and open-reference Greengenes observation tables. Note that Deblur removes approximately one-third to one-half of sequences owing to suspected errors, which is consistent with a sequence length of ~90–150 bp and an average error rate of 0.006 per position62.
Comparison against reference databases
To compare the unique sequence diversity in this study to that in existing databases, sequences from the complete Deblur 90-bp observation table were compared to the set of unique full-length sequences from Greengenes 13.8 and the non-eukaryotic fraction of Silva 128 databases using the open-source sequence search tool VSEARCH63 in global alignment search mode, requiring 100% similarity across the query sequence and allowing multiple 100% reference matches.
Prevalence as a function of sequencing depth
The QC-filtered Deblur 90-bp observation table was additionally filtered to samples that had at least 50,000 sequences (observations). We chose to focus on four environment types (EMPO level 3) where there were many hundreds of samples with more than 50,000 sequences: soil (n = 2,279), saltwater (n = 478), freshwater (n = 1,508), and animal distal gut (n = 695) environments. For each environment, the observation tables were randomly subsampled to 50, 500, 5,000, and 50,000 sequences per sample. The prevalence of each tag sequence was determined as the number of non-zero occurrences across samples divided by the total number of samples. We then plotted a histogram of tag sequence prevalence at each sampling depth. In order to control for potential study bias, we ran the same analysis on a subset of the observation tables where 30 samples were randomly sampled from each study (studies with fewer than 30 samples with > 50,000 sequences were discarded). To investigate how mean tag sequence prevalence changes with increasing sequencing depth across environments, we calculated the average mean tag sequence prevalence across three replicate rarefactions. We plotted the average and standard deviation in mean prevalence across replicate subsamples over a subsampling gradient (that is, 50, 100, 500, 1,000, 5,000, 10,000 and 50,000 sequences per sample).
Greengenes insertion tree
Deblur tag sequences were inserted into the Greengenes reference tree using SEPP55, which uses a divide-and-conquer technique to enable phylogenetic placement on very large reference trees. The SEPP method uses HMMER64 internally for aligning each Deblurred sequence to a reference Greengenes alignment (gg_13_5_ssu_align_99_pfiltered.fasta) with 99% threshold for clustering (resulting in 203,452 tag sequences) and dividing the reference alignment to subsets with a thousand sequences each. It then uses pplacer65 to insert the sequences into the reference Greengenes tree (99_otus.tree), dividing it into subsets of size 5,000. The branch lengths on the Greengenes tree were recomputed using RAxML66 under the GTRCAT model before the placement. The pipeline used, including the reference trees and alignments can be found at ftp://ftp.microbio.me/emp/release1/otu_info/greengenes_sepp_pipeline, and the bash script is available at https://github.com/biocore/emp/blob/master/code/03-otu-picking-trees/deblur/run_sepp.sh.
Fast UniFrac
Unweighted and weighted UniFrac were computed using the Cythonized67 implementation of Fast UniFrac54 in scikit-bio68. Fast UniFrac by itself was not scalable for the EMP dataset owing to an intermediary data structure required by the algorithm, which scales in space by O(NM), where N is the number of nodes in the phylogeny and M is the number of samples. A workaround was designed and implemented in scikit-bio (skbio.diversity.block_beta_diversity) which computes partial distance matrices as opposed to all samples pairwise, enabling large reductions within the intermediary data structure by shrinking M and, in tandem, shrinking N to only the relevant nodes of the phylogeny. This decomposition also allows a classic map-reduce parallel approach with low per-process space requirements. Further space and time reductions were obtained through the implementation and use of a balanced-parentheses tree representation69 (https://pypi.python.org/pypi/iow).
Core diversity analyses: alpha- and beta-diversity
Alpha-rarefaction was computed using single_rarefaction.py in QIIME 1.9.160 using as input the Deblur 90-bp BIOM table and rarefaction depths of 1,000, 5,000, 10,000, 30,000, and 100,000. Alpha-diversity was computed using scikit-bio 0.5.0 with the input Deblur 90-bp BIOM table rarefied to 5,000 observations per sample, and alpha-diversity indices were observed_otus (number of unique tag sequences), shannon (Shannon diversity index70), chao1 (Chao 1 index71), and faith_pd (Faith’s phylogenetic diversity72, using the Greengenes insertion tree). Fast UniFrac54 was run on the Deblur 90-bp table using the aforementioned approach and the corresponding insertion tree. Principal coordinates were computed using QIIME 1.9.1.
Effect size calculations of alpha- and beta-diversity
A version of the mapping file (metadata) was compiled containing the predictors to be tested: study_id, host_scientific_name (a proxy for host taxonomy), latitude_deg, longitude_deg, envo_biome_3 (a proxy for biome or environment), empo_3 (a proxy for sample type or environment generally), temperature_deg_c, ph, salinity_psu, and nitrate_umol_per_l (a proxy for nutrient levels generally). Predictors chosen were those expected to be less redundant with other predictors not chosen, with the exception that there was substantial overlap between study ID and many of the other predictors—because independent studies typically focused on limited sample types from constrained geographic ranges, it is expected that study ID serves as a proxy for a wide range of other measured and unmeasured environmental variables (see Extended Data Fig. 5b). Categories for each predictor were chosen as follows: numerical data were first converted to categories using quartiles; then each category was required to be found in at least 0.3% of all samples (corresponding to 75 samples); categories that were less common than this were ignored. Note that some predictors in our data have complex nonlinear relationships that multivariate statistical analyses using quartiles may miss, such as the unimodal upper-constraint-based richness relationships of temperature and pH. We then tested the effect size of each predictor versus the number of observed tag sequences (alpha-diversity) and weighted and unweighted UniFrac distances (beta-diversity). Effect size was calculated using a Python implementation of the mixed-directional false discovery rate (mdFDR)73,74. mdFDR reduces the false discovery rates by penalizing the multiple pairwise comparisons within each metadata category and the multiple metadata category comparisons. mdFDR has four steps. First, it performs a pairwise comparison (Mann–Whitney U for alpha-diversity, and PERMANOVA for beta-diversity) of each group within each category. Second, for each category we calculate a pooled P value based on the P values of all pairwise comparisons for any given category. Third, we apply the Benjamini–Hochberg procedure to the pooled P values and remove non-significant metadata categories. Finally, we estimate the effect size of those categories found to be significant in step 3 and that have a pairwise comparison P value greater than (R/m × qi) × α, where R is the number of categories that were found significant, m is the number of categories that are being compared (the original number of categories in the input mapping file), qi is the number of pairwise comparisons in each given category, and α is the control level for FDR. The effect size for a given metadata column is calculated as the difference of means of each pairwise comparison divided by pooled standard deviation. To further assess the combined effect size of predictors with non-redundant explanatory power on alpha- and beta-diversity, the non-redundant predictors were selected by forward stepwise redundancy analysis with the R package vegan75 ordiR2step function. This analysis provides an estimate of the relative contribution of each non-redundant predictor to the combined effect size and their independent fraction to the community variation.
Average community 16S rRNA gene copy number
The closed-reference observation table (Greengenes 13.8) was run through the PICRUSt 1.1.0 command normalize_by_copy_number.py script76, which divides the abundance of each OTU by its inferred 16S rRNA gene copy number (that is, copy number is inferred from the closest genome representative for a Greengenes 16S rRNA gene reference sequence). Samples with more than 10,000 sequence reads were summed (that is, OTU abundances were summed within each sample) in both the copy-number normalized and original observation tables. The weighted average community 16S rRNA gene copy number (ACN) for each sample was calculated as the raw sample sum divided by the normalized sample sum.
Covariation of richness with latitude, pH, and temperature
Measurements of alpha-diversity were compared to absolute latitude using a linear mixed-effects model incorporating study ID as a random variable and the interaction of environment and latitude as fixed effects; this was performed on a dataset filtered to include only studies comprising samples that spanned at least 10° of absolute latitude. Correlation of richness with pH and temperature were fitted with a Laplace distribution. The Laplace distribution is a continuous probability distribution that simultaneously captures exponential increase and exponential decrease around a modal value (μ). This distribution is also referred to as the double exponential or two-sided exponential because it represents two symmetrical exponential distributions back-to-back. The Laplace is particularly useful for testing the biological hypothesis that a system is under strong selection to take a particular value (μ) and that small deviations from μ produce an exponential decrease, for example, in diversity. We tested this hypothesis with regards to how tag sequence richness (S) relates to pH and temperature. We used the upper 99th percentile of tag sequence richness across narrow ranges of pH (100 bins) and temperature (120 bins), meaning that our question pertained to the relationship of maximum tag sequence richness (Smax) to pH and temperature. We compared our expectations of exponential decrease in maximum S against the fit to a Gaussian curve, which can also predict a steep symmetrical decrease with small deviations from μ.
Random forest classification of samples
Random forest classification models were trained on the 2,000-sample subset of the Deblur 90-bp observation table to test classification success of samples into the environmental categories from which they came. The R packages caret77 and randomForest78 were used. Five repeats of tenfold cross-validation were used to evaluate the classification accuracy. Confusion matrices were computed to measure the agreement between prediction and true observation. The models were then used to classify the other remaining samples in the full QC-filtered subset.
SourceTracker analyses
SourceTracker79 uses a Bayesian classification model together with Gibbs sampling to predict the proportion of tag sequences from a given set of source environments that contribute to sink environments. We applied SourceTracker 2.0.1 (http://github.com/biota/sourcetracker2) to define the degree to which tag sequences are shared among environmental samples, using the 2,000-sample subset of the Deblur 90-bp observation table (~20% of each sample type) as source samples to train the model, and the remainder as sink samples to test the model. Additionally, we used leave-one-out cross-validation to predict the sample type of each source sample when that sample type is excluded from the model, in order to evaluate the homogeneity of source samples and independence of each source type. Source and sink samples were rarefied to 1,000 sequences per sample before feature selection and testing.
Nestedness of taxonomic composition
Nestedness captures the degree to which elements of a large set are contained within progressively smaller sets. We used the NODF statistic80 to quantify nestedness of the sample-by-taxa matrix. The rows of this matrix correspond to specific taxa grouped at particular taxonomic levels (for example, phylum, class, etc.), while the columns correspond to particular samples. After sorting the matrix from greatest-to-least according to row and column sums, we quantified two aspects of the NODF statistic. The first is a ‘row’ version of NODF that quantifies the degree to which ranges of less prevalent taxa are subsets of the ranges of more prevalent taxa. The second is a ‘column’ version of NODF that quantifies the degree to which less diverse communities are subsets of more diverse communities. We employed two null models to better interpret the observed values of the NODF statistic. The first is based on a random shuffling of occurrences within each row, holding row sums constant (fixed rows, equiprobable columns), while the second is based on a random shuffling of occurrences within each column, holding column sums constant (equiprobable rows, fixed columns)81. The results from both of these null models were qualitatively consistent, so we only report findings using the equiprobable rows, fixed columns model, as it is more consistent with rarefaction of the observation tables. We considered null models at each taxonomic level (phylum, class, order, family, genus), and for all of the samples and each subset of the samples at EMPO level 2. To compute standardized effect scores (SES), we used analytical results based on the hypergeometric distribution to find the expectation and variance of the NODF statistic under both models. SES values were generally very large (>2); we used Wald tests to compute approximate P values.
Environment distribution of taxa and Shannon entropy
For each Deblur tag sequence B, sample s in the set of all EMP samples S, and sample type (EMPO level 3) category E, define

as the fraction of total appearances of tag sequence B in sample type category E (with N possible values). For a given cluster of tag sequences T (phylogenetic subtree or taxonomic group, for example, Firmicutes), we then calculate cluster distribution vector as

where WE combined for all tag sequences in the sequence cluster is given by

Clusters of tag sequences were defined in two ways: first, by partitioning using the taxonomic lineage information for the tag sequences; second, by maximum tip-to-tip branch length for nodes on the phylogenetic tree. To calculate entropy of environment distribution as a function of taxonomic level (for example, phylum), the mean of Shannon entropies for all taxonomic groups belonging to that taxonomic level was calculated (weighted by the number of tag sequences in each taxonomic group). To calculate the entropy as a function of the phylogenetic subtree group width, cluster Shannon entropy was calculated for all subtrees, as well as the maximum tip-to-tip distance for each subtree. To ascertain whether changes in entropy between taxonomic and phylogenetic levels were expected given the observed distribution of environment entropy among tag sequences, a null model was calculated by randomly permuting the Deblur tag sequence taxonomy associations (for the entropy versus taxonomy analysis) or the phylogenetic tip placement (for the entropy versus phylogeny analysis). To reduce the effect of discretization on the entropy calculation in both analyses, clusters of tag sequences were included in the analysis only if they had a minimum of 20 tag sequences. For unique tag sequences (that is, a branch length threshold of 0.0), sequences were required to be found in a minimum of 10 samples. To calculate the approximate branch length corresponding to each taxonomic level, we found the lowest common ancestor for each group and calculated the maximum tip-to-tip distance in that subtree.
EMP trading cards
We started with a BIOM table of 90-bp Deblur tag sequences (16S rRNA gene, V4 region), rarefied to 5,000 observations per sample, containing 2,000 samples evenly distributed across environments and studies (Extended Data Fig. 7a). From this we calculated the following: the number, fraction, and rank of samples in which a tag sequence is found; the abundance, fraction, and rank of observations represented by that tag sequence; the taxonomy of the tag sequence from Greengenes; and the list of all the samples in which the tag sequence is found. This summary is located at ftp://ftp.microbio.me/emp/release1/otu_distributions/. Additionally, for each tag sequence with a trading card in Extended Data Fig. 7b or http://www.earthmicrobiome.org/trading-cards, we identified sequences in RDP (http://rdp.cme.msu.edu)82 matching 100% along the 90-bp region of the 16S rRNA gene. Trading cards at http://www.earthmicrobiome.org/trading-cards are those with prevalence or abundance in the top 10 of all tag sequences or the most abundant tag sequence for each environment having a distribution Shannon entropy < 1, a proportion of that environment ≥ 25%, and total observations ≥ 1,000.
Redbiom database service
A metadata and feature search service containing the EMP data is available through Redbiom. Redbiom is a caching layer for BIOM table and sample metadata, where by default it allows users to interact with the public portion of Qiita (which includes all of the EMP studies). This service allows users to find samples on the basis of sample details (for example, all soil samples with pH < 7), to find samples on the basis of features they contain (for example, all samples in which Greengenes ID 131337 exists), to find features on the basis of taxonomy (for example, all samples in which genus Pyrobaculum exists), to extract sample data into BIOM tables, and to extract sample metadata. Installation of the command-line client and usage instructions are available at https://pypi.python.org/pypi/redbiom; examples of command-line queries are provided at https://github.com/biocore/redbiom. A graphical user interface for Redbiom is available at http://qiita.microbio.me.
Code availability
Code for reproducing sequence processing, data analysis, and figure generation is provided at https://github.com/biocore/emp and is archived at https://zenodo.org with DOI 10.5281/zenodo.1009693. Redbiom code is available at https://github.com/biocore/redbiom and is archived at https://zenodo.org with DOI 10.5281/zenodo.1009150.
Data availability
Per-study sequence files and sample metadata are available from EBI (http://www.ebi.ac.uk/ena) with accession numbers in Supplementary Table 1. Per-study sequence files, sample metadata, and observation tables and information are available from Qiita (http://qiita.microbio.me) using the study IDs in Supplementary Table 1. EMP-wide sample metadata, observation tables and information (trees and taxonomies), alpha- and beta-diversity results, and observation summaries for trading cards are available at ftp://ftp.microbio.me/emp/release1; these files plus the Redbiom database at time of publication are archived at https://zenodo.org with DOI 10.5281/zenodo.890000.
Change history
04 December 2017
The hyperlink “https://qiita.microbio.me” was corrected to “http://qiita.microbio.me”.
References
Lozupone, C. A. & Knight, R. Global patterns in bacterial diversity. Proc. Natl Acad. Sci. USA 104, 11436–11440 (2007)
Ley, R. E., Lozupone, C. A., Hamady, M., Knight, R. & Gordon, J. I. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788 (2008)
Fierer, N. & Jackson, R. B. The diversity and biogeography of soil bacterial communities. Proc. Natl Acad. Sci. USA 103, 626–631 (2006)
Steele, J. A. et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 5, 1414–1425 (2011)
Philippot, L., Raaijmakers, J. M., Lemanceau, P. & van der Putten, W. H. Going back to the roots: the microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799 (2013)
Lima-Mendez, G. et al. Ocean plankton. Determinants of community structure in the global plankton interactome. Science 348, 1262073 (2015)
Gilbert, J. A . et al. Meeting report: the terabase metagenomics workshop and the vision of an Earth microbiome project. Stand. Genomic Sci. 3, 243–248 (2010)
Gilbert, J. A., Jansson, J. K. & Knight, R. The Earth Microbiome project: successes and aspirations. BMC Biol. 12, 69 (2014)
Shade, A., Caporaso, J. G., Handelsman, J., Knight, R. & Fierer, N. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J. 7, 1493–1506 (2013)
Yilmaz, P. et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 29, 415–420 (2011)
Buttigieg, P. L., Morrison, N., Smith, B., Mungall, C. J. & Lewis, S. E. The environment ontology: contextualising biological and biomedical entities. J. Biomed. Semantics 4, 43 (2013)
Olsen, G. J., Lane, D. J., Giovannoni, S. J., Pace, N. R. & Stahl, D. A. Microbial ecology and evolution: a ribosomal RNA approach. Annu. Rev. Microbiol. 40, 337–365 (1986)
Goodwin, K. D. et al. DNA sequencing as a tool to monitor marine ecological status. Front. Mar. Sci. https://doi.org/10.3389/fmars.2017.00107 (2017)
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl Acad. Sci. USA 108 (Suppl. 1), 4516–4522 (2011)
Eloe-Fadrosh, E. A., Ivanova, N. N., Woyke, T. & Kyrpides, N. C. Metagenomics uncovers gaps in amplicon-based detection of microbial diversity. Nat. Microbiol. 1, 15032 (2016)
Hug, L. A. et al. A new view of the tree of life. Nat. Microbiol. 1, 16048 (2016)
Apprill, A., McNally, S., Parsons, R. & Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137 (2015)
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016)
Walters, W. et al. Improved bacterial 16S rRNA gene (V4 and V4–5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 1, e00009–15 (2016)
Sogin, M. L. et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl Acad. Sci. USA 103, 12115–12120 (2006)
Amir, A. et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2, e00191–16 (2017)
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. http://dx.doi.org/10.1038/ismej.2017.119 (2017)
Huse, S. M., Welch, D. M., Morrison, H. G. & Sogin, M. L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12, 1889–1898 (2010)
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 87 (2014)
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618 (2012)
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013)
Nemergut, D. R. et al. Decreases in average bacterial community rRNA operon copy number during succession. ISME J. 10, 1147–1156 (2016)
Gibbons, S. M. et al. Invasive plants rapidly reshape soil properties in a grassland ecosystem. mSystems 2, e00178–16 (2017)
Klappenbach, J. A., Dunbar, J. M. & Schmidt, T. M. rRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 66, 1328–1333 (2000)
Hillebrand, H. On the generality of the latitudinal diversity gradient. Am. Nat. 163, 192–211 (2004)
Fuhrman, J. A. et al. A latitudinal diversity gradient in planktonic marine bacteria. Proc. Natl Acad. Sci. USA 105, 7774–7778 (2008)
Ladau, J. et al. Global marine bacterial diversity peaks at high latitudes in winter. ISME J. 7, 1669–1677 (2013)
Milici, M. et al. Low diversity of planktonic bacteria in the tropical ocean. Sci. Rep. 6, 19054 (2016)
Chu, H. et al. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ. Microbiol. 12, 2998–3006 (2010)
Wu, Y., Zeng, J., Zhu, Q., Zhang, Z. & Lin, X. pH is the primary determinant of the bacterial community structure in agricultural soils impacted by polycyclic aromatic hydrocarbon pollution. Sci. Rep. 7, srep40093 (2017)
Zhou, J. et al. Temperature mediates continental-scale diversity of microbes in forest soils. Nat. Commun. 7, 12083 (2016)
Hendershot, J. N., Read, Q. D., Henning, J. A., Sanders, N. J. & Classen, A. T. Consistently inconsistent drivers of microbial diversity and abundance at macroecological scales. Ecology 98, 1757–1763 (2017)
Carvalho, J. C., Cardoso, P., Borges, P. & Schmera, D. Measuring fractions of beta diversity and their relationships to nestedness: a theoretical and empirical comparison of novel approaches. Oikos 122, 825–834 (2013)
Sonnenburg, E. D. et al. Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215 (2016)
Atmar, W. & Patterson, B. D. The measure of order and disorder in the distribution of species in fragmented habitat. Oecologia 96, 373–382 (1993)
Lomolino, M. V. Investigating causality of nestedness of insular communities: selective immigrations or extinctions? J. Biogeogr. 23, 699–703 (1996)
Gaston, K. & Blackburn, T. Pattern and Process in Macroecology (Wiley-Blackwell, 2000)
Pointing, S. B., Fierer, N., Smith, G. J. D., Steinberg, P. D. & Wiedmann, M. Quantifying human impact on Earth’s microbiome. Nat. Microbiol. 1, 16145 (2016)
Dornelas, M. et al. Assemblage time series reveal biodiversity change but not systematic loss. Science 344, 296–299 (2014)
Amano, T., Lamming, J. D. L. & Sutherland, W. J. Spatial gaps in global biodiversity information and the role of citizen science. Bioscience 66, 393–400 (2016)
Ioannidis, J. P. A. The mass production of redundant, misleading, and conflicted systematic reviews and meta-analyses. Milbank Q. 94, 485–514 (2016)
Davies, N. et al. The founding charter of the Genomic Observatories Network. Gigascience 3, 2 (2014)
Alivisatos, A. P. et al. MICROBIOME. A unified initiative to harness Earth’s microbiomes. Science 350, 507–508 (2015)
Gohl, D. M. et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 34, 942–949 (2016)
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012)
Buttigieg, P. L., Pafilis, E. & Lewis, S. E. The Environment Ontology in 2016: bridging domains with increased scope, semantic density, and interoperation. J. Biomed. Semant. 7, 57 (2016)
McDonald, D. et al. The Biological Observation Matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. Gigascience 1, 7 (2012)
Rideout, J. R. et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2, e545 (2014)
Hamady, M., Lozupone, C. & Knight, R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 4, 17–27 (2010)
Mirarab, S., Nguyen, N. & Warnow, T. SEPP: SATé-enabled phylogenetic placement. Pac. Symp. Biocomput. 2012, 247–258 (2012)
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2, 16 (2013)
Mungall, C. J., Torniai, C., Gkoutos, G. V., Lewis, S. E. & Haendel, M. A. Uberon, an integrative multi-species anatomy ontology. Genome Biol. 13, R5 (2012)
Cooper, L. et al. The plant ontology as a tool for comparative plant anatomy and genomic analyses. Plant Cell Physiol. 54, e1 (2013)
Chibucos, M. C. et al. An ontology for microbial phenotypes. BMC Microbiol. 14, 294 (2014)
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010)
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012)
Schloss, P. D., Gevers, D. & Westcott, S. L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6, e27310 (2011)
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016)
Eddy, S. R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009)
Matsen, F. A., Kodner, R. B. & Armbrust, E. V. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11, 538 (2010)
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014)
Behnel, S. et al. Cython: the best of both worlds. Comput. Sci. Eng. 13, 31–39 (2011)
Rideout, J. R. et al. scikit-bio: scikit-bio 0.5.0: Python 3 only release (2016)
Cordova, J. & Navarro, G. Simple and efficient fully-functional succinct trees. Theor. Comput. Sci. 656, 135–145 (2016)
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423 (1948)
Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 11, 265–270 (1984)
Faith, D. P. Conservation evaluation and phylogenetic diversity. Bio. Conserv. 61, 1–10 (1992)
Guo, W., Sarkar, S. K. & Peddada, S. D. Controlling false discoveries in multidimensional directional decisions, with applications to gene expression data on ordered categories. Biometrics 66, 485–492 (2010)
Grandhi, A., Guo, W. & Peddada, S. D. A multiple testing procedure for multi-dimensional pairwise comparisons with application to gene expression studies. BMC Bioinformatics 17, 104 (2016)
Oksanen, J. et al. vegan: Community Ecology Package R package version 2.4-3 (2017)
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013)
Kuhn, M. Building predictive models in R using the caret package. J. Stat. Soft. 28, 1–26 (2008)
Liaw, A. & Wiener, M. Classification and regression by randomForest. R News 2–3, 18–22 (2002)
Knights, D. et al. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–763 (2011)
Almeida-Neto, M., Guimaraes, P., Guimaraes, P. R. J., Loyola, R. D. & Ulrich, W. A consistent metric for nestedness analysis in ecological systems: reconciling concept and measurement. Oikos 117, 1227–1239 (2008)
Ulrich, W., Almeida-Neto, M. & Gotelli, N. J. A consumer’s guide to nestedness analysis. Oikos 118, 3–17 (2009)
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642 (2014)
Arons, M. S., Fernando, L. & Polayes, I. M. Pasteurella multocida--the major cause of hand infections following domestic animal bites. J. Hand Surg. Am. 7, 47–52 (1982)
Ormerod, K. L. et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 4, 36 (2016)
Acknowledgements
We thank J. DeReus for management of information systems; J. Huntley and K. Jepsen for management of sequencing facilities; B. Erickson for administrative assistance; J. Lennon for discussions about macroecological theory; S. Peddada for assistance with effect size calculations; P. L. Buttigieg, C. Mungall, and D. Siegele for assistance with ontologies; A. Rose, A.-S. Roy, A. Bearquiver, B. Cohen, C. Tischer, C. Feh, D. Winkler, E. Jones, E. Angert, F. Blackwolf, G. Martin, H. Schunck, K. Hallinger, L. R. McGuinness, M. Mühling, M. Lombardo, R. Madsen, S. Bowatte, S. Romac, S. Garcia-Houchins, V. Harriman, and W. James for assistance with sample and/or metadata collection; and the following individuals for supporting the project’s founding: A. Scyzrba, A. McHardy, A. Teske, A. Wilke, C. T. Brown, C. Brown, D. Huson, D. Field, D. Evers, D. Wendel, E. Glass, E. Kolke, F. Sun, F. O. Glöckner, G. Kowalchuk, H.-P. Klenk, J. Tiedje, J. Gordon, J. Raes, J. Knight, J. Kostka, J. Heidelberg, J. Eisen, K. E. Wommack, K. Docherty, K. Keegan, K. Konstantindis, M. Bailey, M. Sullivan, N. Desai, N. Kyprides, N. Pace, P. Balaji, R. Gallery, R. Mackelprang, R. O’Dor, R. Ley, T. Vogel, T. Chen, and W. Feng. This work was supported by the John Templeton Foundation (grant ID 44000, Convergent Evolution of the Vertebrate Microbiome), the W. M. Keck Foundation (DT061413), Argonne National Laboratory (US Department of Energy contract DE-AC02-06CH11357), the Australian Research Council, the Tula Foundation, the Samuel Lawrence Foundation, and the Extreme Science and Engineering Discovery Environment (XSEDE, project number BIO150043), which is supported by National Science Foundation grant number ACI-1053575. Funding for L.R.T. was provided in part by NOAA’s Atlantic Oceanographic and Meteorological Laboratory (AOML) and the Mississippi State University/NOAA Northern Gulf Institute. We thank MO BIO Laboratories, Luca Technologies, Eppendorf, Boreal Genomics, Illumina, Roche, and Integrated DNA Technologies for in-kind support at various phases of the project.
Author information
Authors and Affiliations
Consortia
Contributions
J.A.G., J.K.J., and R.K. conceived the idea for the project. L.R.T. coordinated the meta-analysis, performed analysis, and wrote the manuscript. D.M. developed tools, performed analysis, and wrote the manuscript. J.G.S., J.La., K.J.L., R.J.P., S.M.G., A.A., A.T., Z.Z.X., N.A.B., and A.S. performed analysis and wrote the manuscript. Y.V.-B., J.T.M., and S.M. developed tools and performed analysis. A.G. managed the project and performed analysis. J.A.N.-M., S.J.S., E.K., M.F.H., T.K., S.J., L.J., C.J.B., J.Le., Q.Z., J.K., and K.S.P. performed analysis. G.H. and G.A. managed the project. S.M.O., J.H.-M., and D.B.-L. managed the project and coordinated DNA sequencing. K.D.G., R.L.S., A.C., J.A.F., and V.M. wrote the manuscript. N.F., J.K.J., J.A.G., and R.K. managed the project and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks S. Tringe and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Physicochemical properties of the EMP samples.
Pairwise scatter plots of available physicochemical metadata are shown for temperature, salinity, oxygen, and pH, and for phosphate, nitrate, and ammonium. Histograms for each factor are also shown; the number (n) of samples having data for each factor is provided at the top of each histogram. Samples are coloured by environment, and only QC-filtered samples are included. In sample metadata files, environmental factors are named in our recommended format, with analyte name and units combined in the metadata field name.
Extended Data Figure 2 Sequence length, database effects, and beta-diversity patterns.
a, Median sequence length per study after quality trimming. Original EMP studies used 90-bp reads, which were replaced by 100-bp reads for the majority of studies, and have since been replaced by 150–151-bp reads. For most analyses presented in this manuscript, we used the Deblur algorithm and trimmed tag sequences to 90 bp. This allowed inclusion of older studies with shorter read lengths. b, Comparison of Greengenes and SILVA rRNA databases for reference-based OTU picking. Fraction of reads in n = 23,828 biologically independent samples—separated by environment (per-environment n shown in Fig. 1a)—mapping to Greengenes 13.8 and SILVA 123 (97% identity OTUs) with closed-reference OTU picking. Boxplots show median, IQR, and 1.5 × IQR (with outliers). The fraction of reads mapping was similar between Greengenes and SILVA in each environment but slightly higher with SILVA for every environment. c, Alpha-diversity in closed-reference OTUs picked against Greengenes 13.8 and SILVA 123, with sequences rarefied to 100,000, 30,000, 10,000, and 1,000 sequences per sample, displayed as boxplots showing median, IQR, and 1.5 × IQR (with outliers). The sample set for all calculations contained n = 4,667 biologically independent samples having at least 100,000 observations in both Greengenes and SILVA OTU tables. Alpha-diversity metrics were higher with SILVA closed-reference OTU picking than with Greengenes. d, Beta-diversity among all EMP samples using principal coordinates analysis (PCA) of weighted UniFrac distance. Principal coordinates PC1 versus PC2 and PC1 versus PC3 are shown coloured by EMPO levels 2 and 3. As with unweighted UniFrac distance (Fig. 2c), clustering of samples using weighted UniFrac distance could be explained largely by environment.
Extended Data Figure 3 Sequence length effects on observed diversity patterns.
The effect of trimming from 150 bp (the approximate starting length of some sequences) to 90 bp (the trimmed length of all sequences in this meta-analysis) was investigated by comparing alpha- and beta-diversity patterns. All samples, at each sequence length, were rarefied to 5,000 sequences per sample. a, Alpha-diversity distributions of n = 12,538 biologically independent samples displayed as histograms of observed tag sequences coloured by environment (EMPO level 3). Among these samples with sequence length ≥ 150 bp, the distributions are largely preserved when trimming from 150 to 100 to 90 bp. b, Procrustes goodness-of-fit between the 90-bp (grey lines) and 150-bp (black lines) Deblur principal coordinates (unweighted UniFrac distance) for n = 200 randomly chosen samples coloured by environment (EMPO level 2). Beta-diversity patterns between the two sequence lengths are similar.
Extended Data Figure 4 Tag sequence prevalence patterns.
Note that for this meta-analysis, the input observation table was filtered to keep only tag sequences with at least 25 observations total over all samples and then rarefied to 5,000 observations per sample. a, Per-study endemism visualized as a histogram of tag sequences binned by the number of studies in which they are observed (right: linear scale; left: log scale). b, Per-sample endemism visualized as a histogram of tag sequences binned by the number of samples in which they are observed (right: sample counts up to 92 samples and the number of tag sequences in linear scale; left: all tag sequences with bin widths of 100 samples and number of tag sequences in log scale). c, Abundance (total observations in rarefied table) versus prevalence (number of samples observed in) of n = 307,572 tag sequences. Both axes are log scale. The most prevalent tag sequences were also the most abundant. d, Prevalence as a function of sequencing depth in n = 2,279 soil, n = 478 saltwater, n = 1,508 freshwater, and n = 695 animal distal gut samples having at least 50,000 sequences per sample. Shown are the average and s.d. of mean prevalence across triplicate rarefied subsamples of 50, 100, 500, 1,000, 5,000, 10,000, and 50,000 sequences per sample. Average prevalence increases with sequencing depth, and the straight-line relationship on the log–log axis is suggestive of a power law. e, Histograms of tag sequence prevalences at each sampling depth. The histograms show the distribution moving towards higher prevalences with increasing sequencing depth. Gut data lacked tag sequence prevalences > 0.7 owing to the inclusion of very different host species; see f. f, Histograms as in e but on a subset of the observation tables where 30 samples were randomly sampled from each study. Restricting to human gut samples only, the full range of prevalences found in the other environments is observed.
Extended Data Figure 5 Environmental effect sizes, sample classification, and correlation patterns.
a, Effect sizes of predictors on alpha- and beta-diversity. Maximum pairwise effect size (difference between means divided by standard deviation) between categories of each predictor plotted for observed tag sequences (alpha-diversity) and unweighted and weighted UniFrac distance (beta-diversity). Response variables (alpha- and beta-diversity) were derived from the QC-filtered subset of the 90-bp Deblur table containing n = 23,828 biologically independent samples. Numeric predictor variables were converted to quartiles (categorical predictors). Categories within each predictor had a minimum of 75 samples per category. b, Cumulative variation explained by the optimal model of stepwise redundancy analysis (RDA) of predictors: study ID, EMPO level 3, ENVO biome level 3, latitude, and longitude (predictors with values for less than half of samples, including host scientific name, were excluded). Environment (EMPO level 3) and biome (ENVO biome level 3) explained as much variation as study ID when study ID was excluded from the RDA. c, Confusion matrix for random forest classifier of samples to environment (EMPO level 3). The classifier was trained on the 2,000-sample subset, which was then tested on the remaining samples (QC-filtered samples minus 2,000-sample subset). Squares are coloured relative to 100 classification attempts for each true label. Overall success rate was 84%, with the most commonly misclassified sample environments being Surface (non-saline), Animal secretion, Soil (non-saline), and Aerosol (non-saline). d, Receiver operating characteristic (ROC) curve for classification of samples to environment (EMPO level 3). The AUC (area under curve) indicates the probability that the classifier will rank a randomly chosen sample of the given class higher than a randomly chosen sample of other classes. e, Classification success, using a random forest classifier, to EMPO levels 1–3, ENVO material, ENVO feature, and ENVO biome levels 1–3. f, Microbial source tracking: mean predicted proportion of tag sequences from each source environment (EMPO level 3) that occurs in each sink environment. The model was trained on a subset of samples (~20% of each environment), and tested to predict tag sequence source composition in all remaining samples. Aerosol (non-saline), Surface (saline), and Hypersaline samples were not included in this analysis because there were insufficient sample numbers. g, Microbial source tracking: which other environments a sample type most resembles. The model was trained on all source environments except one using a leave-one-out cross-validated model, and then used to classify each sample included in that group. Hence, the predicted classification proportion of environment X to environment X is zero. h, Correlation of microbial richness with latitude. Richness of 16S rRNA tag sequences per sample across EMPO level 2 environmental categories as a function of absolute latitude. Samples from studies that span at least 10° latitude are highlighted in colour, with linear fits displayed per-study as matching coloured lines. Samples from studies with narrower latitudinal origins are shown in grey. The global fit for all samples per category is indicated by a dashed black line.
Extended Data Figure 6 NODF scores of nestedness across samples by taxonomic level.
The NODF statistic represents the mean, across pairs of samples, of the fraction of taxa occurring in less diverse samples that also occur in more diverse samples. A raw NODF of 0.5 would mean that for any pair of samples, on average 50% of the taxa in the less diverse sample would occur in the more diverse sample. a, NODF (raw) and NODF standardized effect size in the 2,000-sample subset by taxonomic level. Results are shown first for all tag sequences and then for tag sequences found in <10%, < 5%, and <1% of samples. By removing the most prevalent tag sequences before analysis (and rarefying only after this step), it was possible to rule out artefacts associated with potential contamination. NODF (raw) is highest at the phylum level and decreases at finer taxonomic levels, and this trend is observed even when the most prevalent tag sequences are removed (removing those occurring in ≥ 10%, ≥ 5%, or ≥ 1% of samples). The decreasing trend is likely to be partially due to finer taxonomic groups having lower prevalence (and lower matrix fill, among other factors) than coarser taxonomic groups, as standardized effect sizes of the NODF statistic are essentially constant across taxonomic levels. b, When five alternate 2,000-sample subsets are randomly drawn (with replacement) from the full (QC-filtered) EMP dataset, the trends in NODF (raw) and NODF standardized effect size remain largely unchanged.
Extended Data Figure 7 Subsets and EMP trading cards.
a, Subsets of the EMP dataset with even distribution across samples and studies. Shown are all EMP samples included in this manuscript (release 1), the QC-filtered subset, and subsets of 10,000, 5,000, and 2,000 samples. The latter three contain progressively more even representation across environments and studies, providing a more representative view of the Earth microbiome and a more lightweight dataset. Top, histograms of samples per environment (EMPO level 3) for each subset. Bottom, histograms of studies per environment (EMPO level 3) for each subset. b, EMP trading cards: distribution of 16S rRNA tag sequences across the EMP. Trading cards highlight the power of the EMP dataset to help define niche ranges of individual microbial sequence types across the planet’s microbial communities. Cards show distribution of 16S rRNA tag sequences in a 2,000-sample subset of the EMP (rarefied to 5,000 observations per sample) having even distribution by environment (EMPO level 3) and study. Taxonomy is from Greengenes 13.8 and Ribosomal Database Project (RDP), with the fraction of exact RDP matches by lineage and species name shown in parentheses. The pie chart and point plot show the relative distribution of environments in which the tag sequence is found (left points) versus the environment distribution of all 2,000 samples (right points). The coloured scatter plots indicate tag sequence relative abundance (normalized to the shared y axis) as a function of metadata values (no points shown indicates that metadata were not provided for that category). For comparison, grey curves with rug plots indicate kernel density estimates of metadata values across all samples in the set of 2,000 (not just samples where the tag sequence was found). Three examples are shown. Left, a prevalent sequence enriched in soil and plant rhizosphere is from the class Acidobacteria, aptly named as this sequence is found at highest relative abundance in low-pH samples. Middle, the sequence most specific for animal surface (also enriched in animal secretion) is annotated as Pasteurella multocida, a common cause of zoonotic infections following bites or scratches by domestic animals, such as cats and dogs83. Right, the sequence most specific for animal proximal gut belongs to S24-7, a family highly localized to the gastrointestinal tracts of homeothermic animals and predominantly found in herbivores and omnivores, but not in carnivores84.
Supplementary information
Supplementary Table 1
This file contains Table 1 - Studies within the Earth Microbiome Project included in this meta-analysis. These studies constitute EMP 16S Release 1. Read length is median bp for sequences in that study. DOIs with an asterisk describe that study’s samples but did not use EMP sequence data. Sample counts exclude controls. (XLSX 91 kb)
Supplementary Table 2
This file contains Table 2 - Description of metadata fields in the mapping files. Also shown is data type (Python/Pandas), variable type, and number of the 27,751 samples having metadata for each field. (XLSX 82 kb)
Supplementary Table 3
This file contains Table 3 - Top tag sequences by prevalence, abundance, and specialization for habitat. The most prevalent (number of samples observed in), abundant (number of total times observed), or specific for a habitat (most abundant tag sequence with prevalence >25% of that habitat and Shannon entropy <1) EMP tag sequences. Tag sequences are 90-bp 16S rRNA gene sequences (V4 region, starting after primer 515f) from the Deblur algorithm. Values are for a subset of the EMP consisting of 2,000 samples with even representation across habitats (EMPO level 3) and studies, except for statistics on blanks, which are for all blank samples in the EMP (values greater in blanks than subset_2k are coloured red). Samples were rarefied to 5,000 observations per sample. tag sequences are sorted by prevalence. Sequences annotated as chloroplast were excluded before statistics were computed. (XLSX 47 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Thompson, L., Sanders, J., McDonald, D. et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463 (2017). https://doi.org/10.1038/nature24621
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature24621
This article is cited by
-
Nutrient availability and plant phenological stage influence the substrate microbiome in container-grown Impatiens walleriana ‘Xtreme Red’
BMC Plant Biology (2024)
-
Biogeographic survey of soil bacterial communities across Antarctica
Microbiome (2024)
-
Differential effects of antiretroviral treatment on immunity and gut microbiome composition in people living with HIV in rural versus urban Zimbabwe
Microbiome (2024)
-
Gut microbiome features and metabolites in non-alcoholic fatty liver disease among community-dwelling middle-aged and older adults
BMC Medicine (2024)
-
A genus in the bacterial phylum Aquificota appears to be endemic to Aotearoa-New Zealand
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
