
Abstract
The characterization of mutational processes that generate sequence diversity in the human genome is of paramount importance both to medical genetics1,2 and to evolutionary studies3. To understand how the age and sex of transmitting parents affect de novo mutations, here we sequence 1,548 Icelanders, their parents, and, for a subset of 225, at least one child, to 35× genome-wide coverage. We find 108,778 de novo mutations, both single nucleotide polymorphisms and indels, and determine the parent of origin of 42,961. The number of de novo mutations from mothers increases by 0.37 per year of age (95% CI 0.32–0.43), a quarter of the 1.51 per year from fathers (95% CI 1.45–1.57). The number of clustered mutations increases faster with the mother’s age than with the father’s, and the genomic span of maternal de novo mutation clusters is greater than that of paternal ones. The types of de novo mutation from mothers change substantially with age, with a 0.26% (95% CI 0.19–0.33%) decrease in cytosine–phosphate–guanine to thymine–phosphate–guanine (CpG>TpG) de novo mutations and a 0.33% (95% CI 0.28–0.38%) increase in C>G de novo mutations per year, respectively. Remarkably, these age-related changes are not distributed uniformly across the genome. A striking example is a 20 megabase region on chromosome 8p, with a maternal C>G mutation rate that is up to 50-fold greater than the rest of the genome. The age-related accumulation of maternal non-crossover gene conversions also mostly occurs within these regions. Increased sequence diversity and linkage disequilibrium of C>G variants within regions affected by excess maternal mutations indicate that the underlying mutational process has persisted in humans for thousands of years. Moreover, the regional excess of C>G variation in humans is largely shared by chimpanzees, less by gorillas, and is almost absent from orangutans. This demonstrates that sequence diversity in humans results from evolving interactions between age, sex, mutation type, and genomic location.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Veltman, J. A. & Brunner, H. G. De novo mutations in human genetic disease. Nat. Rev. Genet. 13, 565–575 (2012)
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013)
Scally, A. & Durbin, R. Revising the human mutation rate: implications for understanding human evolution. Nat. Rev. Genet. 13, 745–753 (2012)
Kong, A. et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012)
Wong, W. S. W. et al. New observations on maternal age effect on germline de novo mutations. Nat. Commun. 7, 10486 (2016)
Goldmann, J. M. et al. Parent-of-origin-specific signatures of de novo mutations. Nat. Genet. 48, 935–939 (2016)
Rebolledo-Jaramillo, B. et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl Acad. Sci. USA 111, 15474–15479 (2014)
Qin, J. et al. The molecular anatomy of spontaneous germline mutations in human testes. PLoS Biol. 5, 1912–1922 (2007)
Aggarwala, V. & Voight, B. F. An expanded sequence context model broadly explains variability in polymorphism levels across the human genome. Nat. Genet. 48, 349–355 (2016)
Koren, A. et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 91, 1033–1040 (2012)
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015)
Chan, K. & Gordenin, D. A. Clusters of multiple mutations: incidence and molecular mechanisms. Annu. Rev. Genet. 49, 243–267 (2015)
Polak, P. et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518, 360–364 (2015)
Rahbari, R. et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 48, 126–133 (2016)
Francioli, L. C. et al. Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 47, 822–826 (2015)
Besenbacher, S. et al. Multi-nucleotide de novo mutations in humans. PLoS Genet. 12, e1006315 (2016)
Yuen, R. K. C. et al. Genome-wide characteristics of de novo mutations in autism. npj Genomic Med. 1, 16027 (2016)
Seplyarskiy, V. B., Andrianova, M. A. & Bazykin, G. A. APOBEC3A/B-induced mutagenesis is responsible for 20% of heritable mutations in the TpCpW context. Genome Res. 27, 175–184 (2017)
Jónsson, H. et al. Whole genome characterization of sequence diversity of 15,220 Icelanders. Sci. Data 4, 170115 (2017)
Roberts, S. A. & Gordenin, D. A. Hypermutation in human cancer genomes: footprints and mechanisms. Nat. Rev. Cancer 14, 786–800 (2014)
Halldorsson, B. V. et al. The rate of meiotic gene conversion varies by sex and age. Nat. Genet. 48, 1377–1384 (2016)
Herbert, M., Kalleas, D., Cooney, D., Lamb, M. & Lister, L. Meiosis and maternal aging: insights from aneuploid oocytes and trisomy births. Cold Spring Harb. Perspect. Biol. 7, a017970 (2015)
Ségurel, L., Wyman, M. J. & Przeworski, M. Determinants of mutation rate variation in the human germline. Annu. Rev. Genomics Hum. Genet. 15, 47–70 (2014)
Amster, G. & Sella, G. Life history effects on the molecular clock of autosomes and sex chromosomes. Proc. Natl Acad. Sci. USA 113, 1588–1593 (2016)
Gao, Z., Wyman, M. J., Sella, G. & Przeworski, M. Interpreting the dependence of mutation rates on age and time. PLoS Biol. 14, e1002355 (2016)
Moorjani, P., Amorim, C. E., Arndt, P. F. & Przeworski, M. Variation in the molecular clock of primates. Proc. Natl Acad. Sci. USA 113, 10607–10612 (2016)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010)
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009)
Costello, M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013)
Kong, A. et al. Parental origin of sequence variants associated with complex diseases. Nature 462, 868–874 (2009)
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010)
Prüfer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014)
Earl, D. et al. Alignathon: a competitive assessment of whole-genome alignment methods. Genome Res. 24, 2077–2089 (2014)
Acknowledgements
We thank all the participants in this study. This study was performed in collaboration with Illumina.
Author information
Authors and Affiliations
Contributions
H.J., F.Z., E.H., M.T.H., K.E.H., E.A.H., and D.F.G. analysed the data. H.J., B.K., S.K., F.Z., E.H., M.T.H., K.E.H., H.P.E., E.A.H., A.G., and D.F.G. created methods for analysing the data. Ad.J., As.J., and O.Th.M. performed the experiments. S.A.G., L.D.W., G.A.A., H.H., T.R., and M.F. collected the samples and information. H.J., P.S., U.T., G.M., A.K., B.V.H., A.H., D.F.G., and K.S. designed the study. H.J., P.S., B.V.H., A.H., D.F.G., and K.S. wrote the manuscript with input from S.N.S., U.T., G.M., and A.K.
Corresponding authors
Ethics declarations
Competing interests
All of the authors are employees of deCODE Genetics/Amgen, Inc.
Additional information
Reviewer Information Nature thanks S. Sunyaev and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Fraction of DNMs of paternal origin.
a, b, Fraction of DNMs from father per proband. c, d, Fraction of DNMs from father per proband against parental age difference at conception (father’s age − mother’s age). e, f, Paternal age at conception against maternal age at conception. In b, d, and f, all probands with phased DNMs were used; meanwhile, in a, c, and e, the analysis was restricted to 225 three-generation probands. The vertical bars in c and d represent 95% confidence intervals using a normal approximation.
Extended Data Figure 2 Absolute mutational spectra as a function of parents’ ages at conception.
a, Phased DNMs from both parents. b, DNMs from mothers. The mutations were aggregated per year, and parent age groups with only one proband were excluded. The numbers of probands per aggregate year were used as weights for linear regression. We restricted results to 225 three-generation probands for this figure. The y axes are different for a and b. The grey areas in a and b represent 95% confidence intervals using a normal approximation.
Extended Data Figure 3 Localized enrichment of the DNM sex ratio.
a, The maternal DNM enrichment in genome-wide context. We contrasted the local DNM sex ratio in each 2-Mb window against the genome-wide average, using a binomial test. The 2-Mb window number is depicted after the chromosome. The vertical line corresponds to log10(0.05/1352). b, DNMs of paternal origin. c, DNMs of maternal origin. The dotted horizontal line is the genome-wide average. All phased DNMs (42,961) were used for this figure.
Extended Data Figure 4 The number of maternal DNMs per window for various subsets.
a, Excluding regions (10,000 bp) with average coverage of <12× or >120× (2-Mb windows). b, Excluding regions (100,000 bp) with false negative rate above 5% (2-Mb windows). c, Restricting to DNMs phased with the three-generation approach (15,746 DNMs, 3Mb windows).
Extended Data Figure 5 The number of maternal DNMs per 2-Mb window without DNMs in segmental duplications or repeat regions.
a, Without DNMs in annotated segmental duplications. b, Without DNMs in annotated repeat regions.
Extended Data Figure 6 The frequency of C>G variants in 1-Mb windows for the 1000 Genomes dataset.
a, Rare variants (<1%). b, Common variants (≥1%). The dotted horizontal line is the genome-wide average.
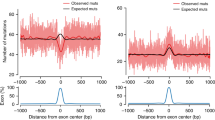
Extended Data Figure 7 Strand concordance and cluster length.
a, Span of multi-nucleotide events in base pairs. b, The concordance of C or G reference bases within multi-nucleotide events as a function of span length. c, Same as b except restricted to C>G DNMs. The vertical bars in b and c represent 95% confidence intervals using a normal approximation.
Extended Data Figure 8 Strand concordance among C>G SNP pairs as a function of genomic position.
a, The concordance ratio. b, The absolute concordance counts.
Extended Data Figure 9 Local C>G enrichment in an evolutionary context for all of the autosomes.
This figure corresponds to Fig. 4c–e for all of the autosomes. a, The C>G divergence between baboon and human, normalized by the C>G divergence between baboon and chimpanzee (db,h/db,c, 1-Mb windows). b, Same as a except the gorilla is used instead of the chimpanzee (db,h/db,g). c, Same as b except the orangutan is used instead of the chimpanzee (db,h/db,o).
Extended Data Figure 10 The phylogenetic context of the dependence of C>G and T>A relative divergence on the rate in the Icelandic dataset.
a, C>G relative divergence against C>G rate. b, T>A relative divergence against T>A rate. The dependency of the relative divergence (C>G or T>A) was modelled against the rate (C>G or T>A) in Icelandic dataset with a linear model. The coefficients of the slopes are reported in the blue rectangles with 95% confidence intervals below the estimates. The dependency of the C>G relative divergence on the C>G SNP patterns in humans reaches its minimum in the ancestral lineage of great apes.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data, Supplementary Tables 1-3, 5-11, 13-20 and Supplementary References. (PDF 605 kb)
Supplementary Table 4
This zipped file contains a gzipped tar archive of the DNMs with proband identifiers. The positions correspond to build hg38 of the human genome. (ZIP 3911 kb)
Supplementary Table 12
This file contains the C>G enriched regions in 1Mb windows. The columns are the following: chromosome, the window number (1Mb) and region. One in the region column correspond to a C>G enriched region. (TXT 24 kb)
Rights and permissions
About this article

Cite this article
Jónsson, H., Sulem, P., Kehr, B. et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549, 519–522 (2017). https://doi.org/10.1038/nature24018
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature24018
This article is cited by
-
Mitochondrial haplotype and mito-nuclear matching drive somatic mutation and selection throughout ageing
Nature Ecology & Evolution (2024)
-
Genetic variation across and within individuals
Nature Reviews Genetics (2024)
-
Assessment of parental mosaicism rates in neurodevelopmental disorders caused by apparent de novo pathogenic variants using deep sequencing
Scientific Reports (2024)
-
Variant in the synaptonemal complex protein SYCE2 associates with pregnancy loss through effect on recombination
Nature Structural & Molecular Biology (2024)
-
Heterozygous loss-of-function variants in DOCK4 cause neurodevelopmental delay and microcephaly
Human Genetics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
