
Abstract
Hundreds of thousands of human genomes are now being sequenced to characterize genetic variation and use this information to augment association mapping studies of complex disorders and other phenotypic traits1,2,3,4. Genetic variation is identified mainly by mapping short reads to the reference genome or by performing local assembly2,5,6,7. However, these approaches are biased against discovery of structural variants and variation in the more complex parts of the genome. Hence, large-scale de novo assembly is needed. Here we show that it is possible to construct excellent de novo assemblies from high-coverage sequencing with mate-pair libraries extending up to 20 kilobases. We report de novo assemblies of 150 individuals (50 trios) from the GenomeDenmark project. The quality of these assemblies is similar to those obtained using the more expensive long-read technology4,8,9,10,11,12,13. We use the assemblies to identify a rich set of structural variants including many novel insertions and demonstrate how this variant catalogue enables further deciphering of known association mapping signals. We leverage the assemblies to provide 100 completely resolved major histocompatibility complex haplotypes and to resolve major parts of the Y chromosome. Our study provides a regional reference genome that we expect will improve the power of future association mapping studies and hence pave the way for precision medicine initiatives, which now are being launched in many countries including Denmark.
Similar content being viewed by others


Main
Using a combination of high-depth (average 78×) Illumina paired-end and mate-pair libraries, we applied Allpaths-LG14 to create de novo assemblies of high quality and coverage for each of the 150 individuals with a median scaffold N50 of ~21 megabases (Mb; maximum ~30 Mb) (Supplementary Table 1). The 100 largest scaffolds in each of the 140 best assemblies typically covered more than 75% (median 77%, Extended Data Fig. 1a) of the genome, with the largest scaffolds exceeding 110 Mb in size (Supplementary Table 1). To evaluate the accuracy of the assemblies, we subsequently aligned the scaffolds for each individual to the human reference genome (GRCh38)15. Figure 1 shows an example individual where the euchromatic part of each chromosome was almost completely covered by a few large scaffolds and in several cases scaffolds covered almost entire chromosome arms. Only rarely did we find that large scaffolds break and align to more than one chromosome (Extended Data Fig. 1b), suggesting that even the largest scaffolds are seldom chimaeric. We also compared our de novo assemblies with a published long-read assembly based on BioNano mapping and PacBio sequencing16. Extended Data Figs 2a and 3 show that this assembly was less complete than our assemblies, but with similar scaffold lengths. The long-read assembly had 5.38% missing data compared with our median of 4.25% (Extended Data Fig. 3a), but the missing data in our assemblies were found in smaller gaps (Extended Data Fig. 3b, c), and the median contig length was therefore much smaller than for the long-read assembly. We conclude that the contiguity of our assemblies is similar to the long-read assembly and second only to the reference genome and the de novo assembly of a hydatidiform haploid mole9,12 (Extended Data Fig. 2b). We identified genomic regions never included in scaffolds >1 Mb (Fig. 1, blue lines) and found them to largely coincide with gaps in the reference genome. They included two known large structural variants found in the reference genome, which were not shared with any of the 100 independent genomes of Danish ancestry. De novo assemblies allowed identification of specific genomic events such as viral integration; for instance, we found telomeric HHV6b integration in one family (Supplementary Figs 1 and 2). In addition to Allpaths-LG, we also assembled the data using SOAPdenovo2 (ref. 16) and SGA17 but found them generally to produce poorer assembly statistics (see Extended Data Fig. 4 and Supplementary Table 2 for details).

For one example individual (983-01), the scaffolds that map to each chromosome are shown (coloured by their rank in length). Most chromosomes are essentially covered by fewer than ten scaffolds; the parts covered by smaller scaffolds are shown as grey. Regions not covered by any scaffolds in any of the 150 assemblies (thin blue line) essentially overlap gaps in the reference genome. The two gaps in the largest scaffolds on chromosomes 6 and 14 correspond to known alternative loci in the reference genome. N regions, regions not assembled in the reference genome.
To fully utilize the de novo assemblies for calling indels and structural variants, and to solve the issue of merging multiple variant call-sets, we devised a hybrid variant calling strategy that first discovered candidate variants on the basis of mapping and assembly, and then genotyped these using a probabilistic method (Fig. 2a). The mapping-based approach used the Genome-Analysis-ToolKit (GATK) with a permissive filter on the recalibrated variant quality scores (99.9% tranche) and identified 22,234,035 candidate variants. In the assembly-based approach, the de novo assemblies were aligned to the GRCh38 reference genome and candidate variants identified using the AsmVar pipeline18, producing a total of 11,469,657 non-single nucleotide variant (non-SNV) candidates. We then genotyped the 150 individuals on the combined set of variants by re-interrogating the raw sequencing data using BayesTyper (J.A.S. et al., manuscript in preparation). The overall trio (mother–father–child) concordance rate (that is, 1 − Mendelian error rate) across all trios and all variants (including structural variants and variants on sex chromosomes) was 98.7% (Extended Data Fig. 5a). After stratifying variants by length and frequency, we randomly selected 200 insertion and deletion variants to be experimentally investigated by cloning, sequencing, and gel electrophoresis. Of these variants, 87.8% were validated, corresponding to an estimated true positive rate of 90.6% (deletions: 95.3%; insertions: 85.7%) after taking into account the number of variants in each stratum (Supplementary Fig. 3). The lower validation rate for insertions was influenced by 5,855 insertions that were large (≥100 nucleotides), of low frequency, and repetitive; the validation rate for insertions increased from 81.6% to 85.5% without these.

a, Candidate alleles were first generated using both mapping-based and assembly-based variant calling pipelines. Permissively filtered candidate alleles and raw sequencing data were then given as input to BayesTyper using a probabilistic model to estimate genotypes. WGS, whole-genome sequencing; LQ, low quality. b, The number of variant alleles called for different types of variation (right) and the proportion of novel calls within each class (left). Bar colours indicate the relative contribution of the two input sources to the total bar width on a linear scale (red, mapping-based; green, assembly-based; yellow, both). Ins, insertions; Del, deletions. c, The variant allele length distribution for insertions and deletions is relatively symmetrical, suggesting that the call-set is not strongly biased towards deletions as in most previous studies; the symmetry also persists for larger structural variants (Extended Data Fig. 5c). Note that insertions with ambiguous bases originating from inter-scaffold gaps were included although their size estimate is associated with some uncertainty (Supplementary Fig. 10). d, e, The proportion of variants that are novel as a function of their size and the proportion of variants containing repeats as a function of their sizes (e), where the peaks around −300 and +300 represent Alu element polymorphisms. SINE, short interspersed nuclear element; nt, nucleotide. f, g, The folded site frequency spectrum shows that the frequency tends to decline with increasing length of the variant allele, whereas the opposite is true for novel structural variants (SV) that are much more likely to be common in the population relative to novel SNVs.
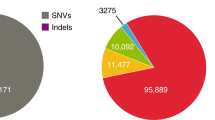
A summary of the final GenomeDenmark call-set is shown in Fig. 2. We found that 16.4% of the called SNVs were novel (not in the Single Nucleotide Polymorphism database 142 (dbSNP142) or 1000 Genomes Project phase 3 structural variant call-set), whereas as many as 91.6% of insertions ≥50 bp were novel (Fig. 2b). The distribution of insertion and deletion calls displayed an excess of insertions (Fig. 2b, c), in contrast to the deletion bias observed in the 1000 Genomes Project and the Genome of the Netherlands19 projects, where our insertion bias partly reflected representational redundancy due to nested variation (Supplementary Fig. 4). The fraction of novel variants increased rapidly with variant length, especially for insertions (Fig. 2d), with most longer variants contributed by the assembly-based approach, which also markedly increased sensitivity for deletions (Extended Data Fig. 5d). For instance, we called 93,052 deletions >20 nucleotides, whereas Genome of the Netherlands called 28,050; we called 33,653 deletions ≥50 bp, whereas the 1000 Genomes Project identified 42,279 such variants in 25 times more individuals who were more diverse than our study population7. Many variants were classified as repetitive, including those responsible for the observed peak in the number of variants with a length between 300 and 350 bases, which could be explained by Alu polymorphisms (Fig. 2e). The allele frequency was observed to decrease with variant length (Fig. 2f), suggesting purifying selection against longer variants20. But looking only at novel variants, we observed the opposite relationship, suggesting that many of the discovered long variants were missed in previous studies (Fig. 2g).
To demonstrate improved sensitivity across the structural variant spectrum more directly, we used our variant calls as a basis for re-genotyping individual NA12878, who was also analysed in the 1000 Genomes Project (Extended Data Fig. 6). We found evidence for the presence of 8,836 of our novel structural variants (≥50 bp; 2,569 deletions and 6,267 insertions), well supported by Mendelian transmission (Extended Data Fig. 6). This directly demonstrates how our data can be used to perform more sensitive genotyping in other sequencing studies. Evaluating protein-truncating variants (PTVs), we found many more indels (59% of the PTVs) compared with the ExAC study (40% of the PTVs)21. Each individual carried, on average, 68 homozygous PTVs (Supplementary Figs 5 and 6) compared with 40 in the ExAC study, with ~24 of these being SNV PTVs in both data sets.
For mutation rate estimations, we identified 3,205 de novo SNVs and 322 de novo indels using GATK, interrogating 2.5 Gb of the genome, with mutation rate estimates for SNVs of 1.28 × 10−8 and for short indels of 1.3 × 10−9 (per generation, generation time 27.7 years). We found many more de novo deletions than insertions and an overrepresentation of even-sized variants (Supplementary Fig. 7, experimental validation rate 92%). Longer de novo indels (>10 bp) were probably under-called using GATK and we further identified 62 of these using BayesTyper. Paternal age was a major determinant of the mutation rate in line with other recent studies11,22,23 (Fig. 3a). Using read-backed phasing, we established the parental origin of 49% of the SNVs and 48% of the indels (Fig. 3b). Significantly more SNVs than indels were of paternal origin (78% versus 66%, P = 0.002), suggesting that these mutations were less dependent on replication. We found a highly significant effect of maternal age on the number of SNV mutations coming from the mother (Fig. 3b), showing that de novo mutations accumulate over time in the female germ line even in the absence of further cell divisions (see also ref. 24). The CpG mutations had a smaller (and non-significant) correlation to paternal age than non-CpG mutations; consequently, the fraction of CpG mutations was negatively correlated with paternal age (Fig. 3c). We also observed significantly higher mutation rates at CpG sites that tended to be methylated in humans (Fig. 3d).

a, The mutation rate per generation as a function of paternal age for SNVs (top) and for indels (bottom). b, Inserted table shows the number of mutations that can be assigned to parental origin for SNVs and indels. The number of mutations received from the father (blue symbols) are correlated with the father’s age and the number of mutations from the mother (red symbols) are correlated with the mother’s age. c, The proportion of mutations that hit CpG sites as a function of paternal age. d, The CpG mutation rate for CpG sites as a function of the methylation rate from H1 cells.
The high-quality de novo assemblies allowed us to resolve full 4–5 Mb major histocompatibility complex (MHC) haplotypes (see Supplementary Fig. 8). We extracted the assembly graph from the scaffolds of each trio and used a combination of alignment within the trio, transmission, remapping against the scaffolds, and read-backed phasing to resolve the individual full MHC haplotypes in 25 of the 50 trios for a total of 100 complete, variable-length MHC haplotypes where >92% of variants were phased (Fig. 4a). These 100 MHC haplotypes complement the pgf haplotype of GRCh38 and the seven previously published haplotypes, where only one (cox) is complete (the other six have 7–49% gaps), bringing the number of complete MHC haplotypes to 102. The accuracy of the haplotypes was supported by validation experiments (86.2% phasing accuracy, Supplementary Table 3). We used unique k-mer stretches to identify a total of 701 kb of novel sequence in the MHC haplotypes (Fig. 4a; see also Methods and Supplementary Table 4). Focusing on a specific example, we found two large fragments of 3 kb and 6.2 kb carried by 22% and 26% of the parental haplotypes, respectively (Fig. 4b). These fragments are absent in the human reference genome (GRCh38) and the IMGT/HLA database, but show high similarity (>98% identity) to a gorilla MHC DRB pseudogene (Gogo-DRBY*01)25. The fragments are flanked by repetitive sequences, including Alu and long interspersed nuclear elements (LINE), which might explain why they have been missed by previous studies.

a, Segments of overlapping novel sequence (identified from 31-mers not found in the reference genome (GRCh38) or the IMGT/HLA database) are plotted along each of the 100 novel MHC haplotypes. Segments are coloured by the length of the segments (log10(length)). b, Length distribution of segments larger than 2 kb (left). Two segments of 3 kb (red) and 6.2 kb (yellow) were common, shared by 35 and 40 individuals (22% and 26% of the parental haplotypes), respectively. The highlighted segments are shown in a subset of the haplotypes that contain both the 3 kb and the 6.2 kb segment and all other segments (>100 bp) in the vicinity (right). The x axis shows the relative position to the centre of the two fragments within each haplotype. c, The Y-chromosome phylogeny is shown along with the number of structural variations for each main branch. The length of the branches is proportional to the number of differences between the individuals. Only structural variants fixed in haplogroups, R1b, R1a, Q1a, I1a, I2a, all I (I1a and I2a), and mutations found in all individuals are shown. Variants are divided into known and novel.
We also fully assembled ~20 Mb of the Y chromosome in long scaffolds (N50 scaffold size over 50 fathers of 1.5 Mb) and found that mainly the very long palindromes and the X-transposed region resisted assembly. We identified 10,898 SNVs, 855 deletions (size range 1–13,620 bp), 793 insertions (size range 1–11,769 bp), and 74 complex variants (size range 1–27,241 bp), and mapped these onto the Y-chromosome tree (Fig. 4c; validation rate 100%, see Supplementary Table 5). We found 181 novel indels fixed in major haplogroups (R, I, and Q) not previously reported26 with a concordance rate of 99.91% (1 father–son mismatch in 1,214 comparisons). We found the mutation rate per generation for structural variants to be 3.26 × 10−9 for deletions and 3.01 × 10−9 for insertions in the ampliconic and X-degenerate regions.
One of the primary goals of the project is to improve interpretation of clinical genetics in Denmark by establishing a regional reference genome. Our extensive catalogue of novel indels and structural variation from the Danish population is tagged well by 1000 Genomes Project SNPs and can therefore be imputed (Extended Data Fig. 7). We also find that there are novel indels in strong linkage disequilibrium with most published findings from genome-wide association studies (GWAS) where the causative variants are not known (Extended Data Fig. 8). As a case example, we investigated whether we could find a higher number of imputed variants in key genomic regions for the Danish GOYA (Genetics of Overweight Young Adults) obesity cohort GWAS (5,222 cases and controls)27.
Combining our panel with the 1000 Genomes Project panel allowed the imputation of an additional 1,204,946 variants compared with the 1000 Genomes Project panel alone and led to a higher accuracy of the imputation, independent of the minor allele frequency (Extended Data Fig. 9). More than a fifth of the additionally imputed variants were insertions and deletions (Extended Data Fig. 9c). These indels improved the coverage of the regions of association in this set, and, for instance, we found that five structural variants were strongly associated with the phenotype and thus candidates for the association on chromosome 16 (see Extended Data Fig. 10).
Exploiting the full potential of the rich resource of structural variants will benefit from genome graph-based methods replacing the use of the single reference genome. Genome graph representations are under development28 and may also represent a solution to the privacy concerns of donors. To illustrate this, we have created and released a fully phased VCF file that is randomly sampled from a probabilistic graph-based data representation retaining most of the linkage disequilibrium structure while respecting donor anonymity (Supplementary Fig. 9).
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Cohort selection
The 50 trios (mother–father–child) were selected from the Copenhagen Family Bank29. A candidate set of 60 trios was selected randomly from a pool of nearly 1,000 while maintaining the constraint of an average Danish male and female height and blood type distribution. The study protocol was reviewed and approved by The Danish National Committee on Health Research Ethics, file number 1210920, submission numbers 36615 and 38259. The HumanCoreExome BeadChip version 1.0 was used to genotype the 60 trios (180 individuals) using the HiScan system (Illumina, San Diego, California, USA). Genotypes were called using GenomeStudio software (version 2011.1; Illumina). All subjects had a high call rate (>98%), and the familial relationship and the sex of the subjects were confirmed. SNPs with a low call rate (<98%) or deviation from Hardy–Weinberg equilibrium (P < 0.0001) were excluded. SNP genotype data from reference populations were obtained from previously published GWAS in Denmark30 and neighbouring populations: that is, Norway31, Sweden32, and Germany33. Standard principal component analysis of the 120 trio parents combined with the GWAS reference data set was conducted after merging data sets and linkage disequilibrium-based pruning of SNPs to assess the homogeneity of the trios and to select a set of 50 trios that best represented the Danish population, thus removing trios with one or more members who appeared as outliers (see Supplementary Fig. 11). From the 60 trios, 7 were removed because of admixed ancestry, shown in the principal component analyses and further confirmed by telephone interview with the families and asking about their ancestry. One trio was removed owing to lack of sufficient blood for sequencing. From the remaining 52 Danish trios, 50 were chosen (final principal component analysis of the 100 parents is shown in Supplementary Fig. 12).
Sequencing and sequence quality control
DNA was extracted from fresh or frozen blood samples of the 150 donors. At least 278 μg was obtained for each individual and used to create seven libraries, with insert sizes 180–230 bp, 500–550 bp, and 750–800 bp for paired-end libraries, and 2 kb, 5 kb 10 kb, and 20 kb for mate-pair libraries. Sequencing was conducted on an Illumina HiSeq2000. SOAPfilter version 2.2 was used to pre-process the sequencing data by filtering reads with adaptor contaminations, reads having more than 40% low-quality bases (Q < 7), or >10% N bases. For mate-pair insert size libraries, reads with erroneous alignment orientations were filtered out (Supplementary Fig. 13 and Supplementary Table 6).
Mapping
All reads from the compendium of libraries were mapped to the human reference genome build 38 (GCA_000001405.15). All paired-end libraries were mapped using BWA-MEM (version 0.7.5a)34 and refined using Stampy (version 1.0.23)35, whereas mate-pair libraries were mapped entirely using Stampy. SAMtools (version 0.1.19)36 was used to process the alignment files and to remove duplicate reads.
Mapping-based variant calling
The Genome-Analysis-ToolKit (GATK) (version 3.2-2)37 was used for variant calling from BAM files. Duplicate marking, base recalibration, and local indel realignment were performed on lane-level BAM files before merging BAM files by sample. We used HaplotypeCaller in the ERC mode to generate the genotype likelihoods for each individual. We combined all variation calls from the 150 individuals and performed joint genotyping. We recalibrated the SNPs and indels separately using the known variant files from GATK bundle 3.2. For SNP recalibration, we used hapmap_3.3, 1000G_omni2.5, and 1000G_phase1.snps as the positive training and true data sets, and dbSNP_v141 as the known data set. For indel recalibration, we used Mills_and_1000G_gold_standard indels as the positive training and true data set, and dbSNP_v141 as the known data set. The metrics ‘DP’, ‘FS’, ‘ReadPosRankSum’, and ‘MQRankSum’ were used in the recalibration process. We decided on the recalibration threshold for both the SNPs and the indels as being 99.0.
De novo assembly
All 150 individuals were individually de novo assembled using the three assemblers SOAPdenovo2 (ref. 16), SGA17, and Allpaths-LG14. Each of the assemblers approaches the de novo assembly problem differently (SGA uses string graphs; SOAPdenovo2 and Allpaths-LG use different implementations of the de Bruijn graph).
For Allpaths-LG (version 51646)14, overlapping paired-end reads with an insert size of 180 nucleotides were added as fragment libraries, and all other libraries (>180 nucleotide insert size) were added as jumping libraries. For one sample, a 5 kb library was discarded as it was error prone and could not be processed by the Allpaths-LG PrepareAllPathsInputs.pl script. Allpaths-LG assemblies were run with default settings, except for ~45 samples that kept failing in a specific module (BuildUnipathLinkGraphsLG). These samples were run with the setting BULG_TRANSITIVE_FILL_IN = False. Tests were performed on different non-failing individuals to assess whether disabling the module would affect the assemblies, but no differences in the assemblies were observed. The SOAPdenovo2 (version r240) assembly of the 150 individuals was performed as done in ref. 2. For SGA (version 0.10.13) the 180 bp paired-end reads were initially collapsed using FLASH (version 1.2.11)38 to single-end reads and thereafter all libraries were processed using the SGA pipeline. Filter was run with –kmer-threshold 2, FM-merge with -m 75, overlap with -m 77, and assemble using -m 77 -d 0.4 -g 0.1 -r 10. Thereafter, contigs were scaffolded iteratively beginning with the smallest library. Scaffolding was performed by mapping with BWA-MEM (version 0.7.12), calculating astat and bam2de, and finally using the scaffold command in SGA.
Assembly-based variant calling
We applied the LAST aligner (version 1.1.0)39 to align the scaffolds to the human reference genome. Split alignment was performed to allow for the existence of genome rearrangements. The misalignment probabilities were computed to provide Phred-scaled confidence measures of the correctness of genome-scale and base-scale alignments. In the final assembly-versus-assembly alignments, every non-overlapping DNA piece of the scaffold was anchored to a unique position in the reference, and we only kept alignments with misalignment probabilities <0.01. Candidate variants were called using the module A in AsmVar (version 1.0.0)18, which detects and characterizes structural variants from the alignments.
Variant integration and genotyping
Variants from mapping-based and assembly-based calling were then integrated and genotyped using BayesTyper 0.9 (J.A.S. et al., manuscript in preparation) on the basis of sample k-mer counts and candidate variants from the two call-sets as input. More specifically, the sample input was obtained by counting the number of occurrences of all 55-mers in the cleaned sequencing data for each individual using KMC2 (version 2.2.0)40 with removal of singleton k-mers enabled. The candidate variants were first filtered by selecting the 99.9 sensitivity tranche of GATK calls and by selecting only non-SNV AsmVar variants that passed the AGE realignment step. The filtered set of variants was then normalized using bcftools norm (version 1.3.1)41 and finally merged using the combine tool from the BayesTyper package. Joint genotyping using the BayesTyper was done in 10 batches of 15 individuals (five parent–offspring trios in each) with the complete variant set from all 150 samples provided as input to each run; all arguments were set to their default values. If not stated otherwise, all post-processing was done using tools and scripts that were part of the BayesTyper package and available at https://github.com/bioinformatics-centre/BayesTyper.
The genotype calls from the 10 batches were combined to create a joint call-set containing the genotypes of all 150 individuals. An allele was classified as called if at least one sample had an allele call posterior greater than or equal to 0.9.
The call-set was further post-processed using sample genotype filters, which were used to handle errors arising from data properties not completely accounted for by the model. In this study, a genotyped allele in a sample was filtered if it was covered by fewer than three observed, informative k-mers. We further filtered homopolymer alternative alleles (across all samples) if they were located in a homopolymer in the reference sequence longer than 15 nucleotides and if they differed only by a single nucleotide in length from another allele. Finally, variants for which the observed number of heterozygotes deviated markedly from the expectation under Hardy–Weinberg equilibrium were filtered by removing variants for which the value |1 − number of observed heterozygotes/number of expected heterozygotes| was larger than 0.8. The filter thresholds were determined empirically by comparing the number of filtered alleles (sensitivity) with the fraction of called annotated alleles. Allele call probabilities were re-estimated after filtering using only the unfiltered sample alleles.
Variant annotation
Each alternative allele was classified as SNV, deletion, insertion, inversion, or complex. Inversions were defined as alternative alleles of equal length to the reference allele, which were at least ten nucleotides long and matching the reverse complement of the reference allele with no more than 5% mismatches. Any variant allele that did not fall into the SNV, deletion, insertion, or inversion categories was labelled as complex.
Each alternative allele in the call-set was annotated using dbSNP142 (dbSNP) and the structural variants from the 1000 Genomes Project phase 3 (1KGSV). Both sets of annotated variants were first normalized using bcftools (version 1.2.1) norm41. Mitochondrial mobile insertions without sequence content were removed from the 1KGSV set before normalization. A custom annotation tool was developed to allow annotation of all variant types (including structural variants) on the basis of sequence overlap. A variant allele was defined as annotated if it matched another variant allele within a window extending three variant lengths up- and downstream of the variant start and end positions, respectively. A match was defined as two variant alleles for which 1 − (edref + edalt)/(max(lengthref1, lengthref2) + max(lengthalt1, lengthalt2)) > 0.5, where ed is the edit distance between either the two reference (ref) or two variant (alt) alleles computed using EdLib in Needleman–Wunsch mode42. All alleles that could not be annotated were classified as novel. The same annotation routine was also used to identify redundant variants by annotating the call-set against itself, but using a window extending only one variant length in each direction and requiring a match score >0.9.
Insertions and deletions were further classified on the basis of repeat content using RepeatMasker. Specifically, the variant sequences (alternative allele sequence for insertions and reference sequence for deletions) were provided as input to RepeatMasker (version 4.0.6) using dfam43 (version 2.0 running HMMER version 3.1b2). A variant was classified as belonging to a certain repeat class if the repeat covered at least 90% of the variant sequence and to be repetitive if the union of sequence intervals covered by repeats covered at least 90% of the sequence.
Validation
To validate the structural variants in the integrated BayesTyper call-set, insertions and deletions that were called as bi-allelic, that contained no ambiguous bases, had no overlap with any other variants, and that were amenable to PCR amplification were selected. For all such variants, a random, heterozygotic sample (with genotype posterior probability > 0.9) was chosen among the 29 trios picked for validation. Primers were then designed for all variants passing this step, and variants for which no primers could be designed were discarded. To create a representative validation set, the variants passing the above criteria were first divided into insertions and deletions. These were then further divided into five different length bins (<5, 6–19, 20–49, 50–99, ≥100) and again into rare and common, with rare alleles defined as having an estimated allele count lower than or equal to 15 (5%). Finally, 10 variants were randomly selected from each bin, providing a total of 200 variants for validation.
Validation was performed by sequencing five cloned PCR products for each variant. For each clone, the forward and reverse sequencing reads were trimmed and a consensus sequence assembled using SeqTrace44 (version 0.9.0) with default parameters; reads for which no consensus sequence could be assembled were discarded. Consensus sequences were aligned to both the reference and alternative alleles including flanks using needle45 with default parameters. A clone was then assigned to an allele if the alignment contained no gaps; clones that could not be assigned to either of the alleles were labelled as invalid. Alignments for all invalid clones were subsequently inspected manually and alleles assigned where possible. The PCR products for all variants longer than 50 nucleotides were also run on a gel and the fragment lengths for each band estimated using GelAnalyzer (version 2010a). An allele was considered validated if it was observed in either the cloning and sequencing-based approach or if its expected size matched that of a band on the gel. The validation rate was computed by dividing the number of alternative alleles that validated by the number of variants for which either the reference or the alternative allele was observed.
We note that the selection of variants for validation was performed on a call-set that was generated using an earlier version of BayesTyper (version 0.8) and a more permissive set of filters. The results shown in Supplementary Fig. 3 were obtained by computing accuracies for the validation variants for the current version of the call-set, which was based on BayesTyper version 0.9 and more stringent filters. We emphasize that, to avoid overfitting to the validation data, these data were not used for deciding any specific changes made to the new version of BayesTyper or for selecting the filter settings.
Re-genotyping NA12878 using the GenomeDenmark call-set
The 50× paired-end and PCR-free Illumina sequencing data for the trio composed of the individuals NA12891 (father), NA12892 (mother), and daughter (NA12878) were obtained from the European Nucleotide Archive (accession number ERP001960). The data were generated as part of the Illumina Platinum Genomes project46, but the data for individual NA12878 were also used to generate the 1000 Genomes Project structural variant calls7. The sample input to BayesTyper was obtained by counting the occurrences of all 55-mers in the sequencing data from the three individuals using KMC2 with counting of singletons enabled. The variation base for genotyping was obtaining by combining unfiltered calls from GATK HaplotypeCaller, Platypus, and FreeBayes for the three individuals obtained from the Illumina Platinum Genomes project (M. Erbele, personal communication, 2016) with the GenomeDenmark call-set and the 1000 Genomes Project structural variant set using the combine function in the BayesTyper package. BayesTyper (version 0.9) was provided with k-mer count tables from the three individuals and the combined set of input variants and then run with default parameters. The output was post-processed as described for the main GenomeDenmark call-set above, but omitting the inbreeding filter owing to the low number of samples. Again, the variants were annotated with variants from dbSNP142 (dbSNP) and structural variants from the 1000 Genomes Project phase 3 (1KGSV).
Calling of PTVs
We used the same pipeline and filters as described in the ExAC study21. We used VEP and LOFTEE, and focused on putative protein-truncating (frameshift, splice donor, splice acceptor, and stop-gained) variants. On top of the stringent filters applied by LOFTEE, we also used the trio information to filter all loci showing Mendelian errors, resulting in 1,495 bi-allelic PTVs (Supplementary Table 7). For comparison, we used the ExAC released data version 0.3.1 (ExAC.r0.3.1.sites.vep.table.gz).
Phasing
We pre-processed the data by setting the trio genotypes with Mendelian error rate as missing and filtered the variants with less than 95% genotyping rate. We subsequently applied Shapeit2 (version 2.r720) to integrate the family relatedness, read linkage, and the linkage disequilibrium information to phase the variations with parameters –assemble, –duo hmm, and –input-map. The genetic map that we used was lifted-over from the b37 version (http://www.shapeit.fr/files/genetic_map_b37.tar.gz) using the University of California, Santa Cruz (UCSC) lift-over script (http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/liftOver) and the hg19ToHg38.over.chain.gz file from UCSC.
After lifting over, we removed all the variants that (1) displayed allele changes, or (2) mapped to more than one location, or (3) had lifted coordinates reversed.
In addition, because the genetic distance changes as the physical distance changes, we recalibrated the genetic distance in the GRCh37 genetic map using the formula m(i) = [p(i) − p(i − 1)] × r(i − 1) + m(i − 1), where m is the genetic distance, p the physical position, and r the recombination rate. Our implementations of the genetic map adopted the approach in the lift-over documentation from the HapMap project (ftp://ftp.ebi.edu.au/pub/software/ensembl/encode/users/anshul/temp/chromatinVariation/rawdata/phasing/geneticMaps/README.txt).
Imputation
We imputed into the GOYA cohort data set27 after lifting over the genotype data to GRCh38 using the ENSEMBL assembly converter. This data set contains 5,222 individuals and 514,705 single nucleotide polymorphisms. The imputation was performed using IMPUTE2 (version 2.3.1) with the lifted-over 1KGP PhaseI and PhaseIII reference panels, the DanishPanGenome reference panel, and the merged panels using the merge option from IMPUTE2. Imputed variants were filtered on the info score generated by IMPUTE2 with a threshold of 0.882. This threshold corresponds to an R2 of 0.8 defined in ref. 47 as a quality cutoff. The R2 score presented in Extended Data Fig. 7 was computed by IMPUTE2.
De novo mutation and SNV calling
We used the approach from our previous study2 with the following filtering criteria: GQ ≥ 80 (for the homozygote filter) and GQ ≥ 250 (heterozygote filter); DP ∈ [20;150] (for both the homozygote and heterozygote filter); AD2 = 4 (for the homozygote filter); allele balance ∈ [0.3; 1] (Supplementary Fig. 14). We also required that the new allele should be seen on both strands.
Parent of origin assignment of de novo mutations
For each variant, X, we used o(X) to denote the parental origin of the alternative allele. The reads might have provided conflicting evidence, so to find the most likely parental origin we calculated a likelihood ratio comparing how likely it was that the alternative allele was on the paternal chromosome (o(X) = 1) with how likely it was that the alternative allele was on the maternal chromosome (o(X) = 0):

If LRX is above 1 it indicates that the alternative allele of variant X is on the paternal chromosome, and if LRX is below 1 it indicates that it is on the maternal chromosome. The data that are informative about the parent of origin are the reads that cover both X and Y:

The probability that a read supports the true phasing is 1 if the read is mapped correctly and ½ if the read is not mapped correctly. We calculated the conditional probability of the read as  where P(rXY correct) is the probability that rXY is mapped correctly (estimated from the phred score in the BAM file) and the values of i and j is either ‘ref’ or ‘alt’ depending on whether the read contained the reference allele or the alternative allele at position X and Y. For inherited variants where the parental origin could be assigned by just looking at the genotypes of the family members, P(o(Y) = 1) was calculated using the Phred-scaled genotype probabilities of the three family members. If the parent of origin of variant Y was assigned using read information, we calculated P(o(Y) = 1) from the estimated LR: P(o(Y) = 1) = LRY/(LRY + 1). The assignment of parental origin was performed iteratively until no additional variants could be assigned.
where P(rXY correct) is the probability that rXY is mapped correctly (estimated from the phred score in the BAM file) and the values of i and j is either ‘ref’ or ‘alt’ depending on whether the read contained the reference allele or the alternative allele at position X and Y. For inherited variants where the parental origin could be assigned by just looking at the genotypes of the family members, P(o(Y) = 1) was calculated using the Phred-scaled genotype probabilities of the three family members. If the parent of origin of variant Y was assigned using read information, we calculated P(o(Y) = 1) from the estimated LR: P(o(Y) = 1) = LRY/(LRY + 1). The assignment of parental origin was performed iteratively until no additional variants could be assigned.
MHC region analysis
Haplotypes of the whole MHC region were constructed using Allpaths-LG scaffolds as the starting point including variants in the fastG version of the scaffolds. Scaffolds aligning to the MHC region in alignment blocks of at least 50 kb were extracted from the assembly graphs. Strand information and median starting points of alignment blocks were used to determine orientation and order of scaffolds to concatenate scaffolds into full-length MHC scaffolds. Scaffolds were trimmed to 1 Mb telomeric of HLA-F and 1 kb centromeric of KIFC1, determined in each case by BLAST48 (blastn, version 2.4.0) of the gene sequences to the MHC scaffolds. Positions of variant sites from the graph were determined within the trio by exact matching 40 bp upstream of each variant. Variants were then phased by transmission. Consensus sequences were created for each parent–offspring haplotype using global alignment between all pairwise sets of phased variants. Haplotypes were refined by first mapping reads to the four haplotypes within each trio using BWA-MEM (version 0.7.5a)34, then calling variants with Platypus (version 0.7.9.1)49, and finally phasing variants that passed quality control by determining the parent of origin of alternative alleles (see above). Gaps in the haplotypes were closed using the GapCloser module from SOAPdenovo2 through five iterations of adding one read library at a time. After gap closing, all transmitted haplotypes were submitted to remapping, variant calling, and phasing as described above. Variant positions in non-transmitted haplotypes were mapped by pairwise alignment to the transmitted haplotypes.
All transmitted haplotypes were aligned to GRCh38 using LAST (version 1.02) aligner39. The AsmVar pipeline was used to create a candidate set of genotypes from the two haplotypes from each individual. BayesTyper was used to call variants from the candidate set of variants, and phasing was restored by using the allele call origin INFO field from AsmVar and removing any variants discordant in respect of phasing and allele call origin. To assess the amount of novel sequence found in the MHC region compared with the reference genome GRCh38 and IMGT/HLA database, we used a k-mer-based approach to detect novel segments. First, a database of all k-mers (k = 31) found in the reference and IMGT/HLA databases was constructed. Then, for each haplotype, we compared the k-mers from that haplotype with the database and kept k-mers not found in the database. We then concatenated all adjacent k-mers to form segments.
To assess the novelty of the sequences, we used RepeatMasker46 to all segments and performed a blastn (version 2.2.26)48 search against the reference genome (GRCh38) including the alternative MHC haplotypes. Conditioning on full query coverage and an identity of at least 98%, we estimated an upper bound for potential non-MHC chimaeric scaffold sequences. We here determine novel MHC sequences as those having no hits, having best hits in the MHC region, or having less than 98% identity in non-MHC parts of the genome. It should be noted that variants falling within distance k = 31 of each other will be detected here as one novel segment instead of two separate events. Highlighting two segments of 3 kb and 6.2 kb that were seen in 23 and 16 haplotypes, respectively, we used BLAST (version 2.2.26) to determine the fraction of individual de novo assemblies carrying these. We blasted against the scaffolds from the de novo assembly of each of the 150 individuals, as we might have missed the fragments in the initial construction of the haplotypes if they were part of scaffolds smaller than 50 kb and to find the fragments in individuals in which we did not construct full MHC haplotypes. Validation was performed as for the BayesTyper variants, for 66 regions including 202 variants.
Y-chromosome analysis
From de novo assemblies, scaffolds aligning to the Y chromosome from the reference (GRCh38) and the LAST (version 1.1.0)49 alignments were extracted. Only scaffolds where the majority of the bases mapped to the Y chromosome with a length >1,000 nucleotides were chosen, to avoid scaffolds that mapped too ambiguously. The gap in the scaffolds was closed with a module from SOAPdenovo called GapCloser (version 1.12)16 and repeat regions were found using RepeatMasker (version 3.3.0)50.
Concordance between father and son was estimated by aligning scaffolds using MAFFT (version 7.245)51 and removing RepeatMasked regions and those 50 bp around alignment gaps. GATK, AsmVar, and BayesTyper were used to identify structural variation18. Variants that were not recurrent (only variable in one haplogroup) were kept for further analysis. Haplotypes were assigned with respect to a minimal list of SNPs52.
The SNVs called using GATK above were used to construct the neighbour-joining tree. The SNVs were required to have a filter status of PASS, not be recurrent, and needed to be in the X-degenerate region. The neighbour-joining tree was constructed using MEGA6 (ref. 53) using the number of substitutions as the model and pairwise deletion as missing data treatment. It was run with 500 bootstrap replicates.
Estimates of the Y-chromosomal substitution rate for complex variation were made on the basis of calibrating our mutation rate of SNVs in X-degenerate regions with the estimate used in ref. 54. Validation was performed on 44 indel variants.
Genome graph implementation
The genome graph is a directed graph consisting of nodes that represent haploid genotypes and edges between pairs of nodes, which indicate genomic adjacency between the genotypes and are annotated with the probability of occurrence of the genotype given its predecessor. This principle is extended into a hypergraph where a hyper-edge may include several consecutive predecessor nodes as well as the successor node. A probability associated with such a hyper-edge reflects the chance of observing the successor node given the specified sequence of predecessor nodes. Tracing a path through the graph corresponds to a particular haplotype; it is used as a stochastic automaton to sample such haplotypes that reflect the chosen edge probabilities. The edge probabilities are derived directly from frequencies of observed genotypes in the 100 parents. If the probability estimate for a hyper-edge is less than 5/100, then it is effectively given probability zero to avoid including individual-specific haplotypes that could in principle be used for identification of individuals. The sampling procedure proceeds by sampling consecutive genotypes where the sampling probability is based on the proceeding sampled genotypes. Generally, the probability-associated largest hyper-edge containing the successor genotype is preferentially used in sampling, but with exponentially decreasing probability a smaller hyper-edge is used instead. This is done to alleviate potential frequency bias introduced by exclusion of rare haplotypes with a support of fewer than five individuals. The sampled haplotypes are finally stored in a phased VCF file, which includes a subset of variants that is compatible with the condition of excluding rare haplotypes described above. The individuals in the resulting VCF are not real individuals, but hypothetical individuals having haplotype frequencies similar the original 100 parents. The genome graph was produced from the phased BayesTyper calls. Pairs of haplotypes were sampled for each 1,000 hypothetical individuals representative of the original 100 parents. Each of the 22 autosomes was processed individually into phased graphs using the vgtool software (https://github.com/Johanvb/VGTool) with default settings.
Data availability
Individual sequence data, alignment based assemblies and the complete variant call-set in the form of a phased VCF file have been deposited at the European Genome-phenome Archive under accession number EGAS00001002108 (https://ega-archive.org/studies/EGAS00001002108). The individual de novo assemblies and MHC haplotypes can be accessed through agreement with the corresponding authors. A population aggregate of the variants has been deposited at the European Variation Archive under accession number PRJEB19794. The full variant call-set can also be freely used for imputation through the International Haplotype Reference Consortium. For unrestricted use, we have released a fully phased VCF file which is randomly sampled from a probabilistic graph-based data representation retaining most of the linkage disequilibrium structure (see Supplementary Fig. 9), while respecting donor consent.
Accession codes
References
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015)
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015)
Hoehe, M. R. et al. Multiple haplotype-resolved genomes reveal population patterns of gene and protein diplotypes. Nat. Commun. 5, 5569 (2014)
Telenti, A. et al. Deep sequencing of 10,000 human genomes. Proc. Natl Acad. Sci. USA 113, 11901–11906 (2016)
Besenbacher, S. et al. Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat. Commun. 6, 5969 (2015)
Sudmant, P. H. et al. Global diversity, population stratification, and selection of human copy-number variation. Science 349, (2015)
Sudmant, P. H. et al. An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81 (2015)
Huddleston, J. et al. Reconstructing complex regions of genomes using long-read sequencing technology. Genome Res. 24, 688–696 (2014)
Steinberg, K. M. et al. Single haplotype assembly of the human genome from a hydatidiform mole. Genome Res. 24, 2066–2076 (2014)
Cao, H. et al. De novo assembly of a haplotype-resolved human genome. Nat. Biotechnol. 33, 617–622 (2015)
Kloosterman, W. P. et al. Characteristics of de novo structural changes in the human genome. Genome Res. 25, 792–801 (2015)
Pendleton, M. et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat. Methods 12, 780–786 (2015)
Seo, J. S. et al. De novo assembly and phasing of a Korean human genome. Nature 538, 243–247 (2016)
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. USA 108, 1513–1518 (2011)
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017)
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012)
Simpson, J. T. & Durbin, R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 22, 549–556 (2012)
Liu, S. et al. Discovery, genotyping and characterization of structural variation and novel sequence at single nucleotide resolution from de novo genome assemblies on a population scale. Gigascience 4, 64 (2015)
The Genome of the Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 46, 818–825 (2014)
Cooper, G. M. et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 43, 838–846 (2011)
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016)
Francioli, L. C. et al. Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 47, 822–826 (2015)
Kong, A. et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012)
Wong, W. S. et al. New observations on maternal age effect on germline de novo mutations. Nat. Commun. 7, 10486 (2016)
Kupfermann, H., Mayer, W. E., O’hUigin, C., Klein, D. & Klein, J. Shared polymorphism between gorilla and human major histocompatibility complex DRB loci. Hum. Immunol. 34, 267–278 (1992)
Poznik, G. D. et al. Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequences. Nat. Genet. 48, 593–599 (2016)
Paternoster, L. et al. Genome-wide population-based association study of extremely overweight young adults - the GOYA study. PLoS ONE 6, e24303 (2011)
Novak, A. M. et al. Genome graphs. bioRxiv http://dx.doi.org/10.1101/101378 (2017)
Eiberg, H. et al. Linkage between serum cholinesterase 2 (CHE2) and gamma-crystallin gene cluster (CRYG): assignment to chromosome 2. Clin. Genet. 35, 313–321 (1989)
Børglum, A. D. et al. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol. Psychiatry 19, 325–333 (2014)
Espeseth, T. et al. Imaging and cognitive genetics: the Norwegian Cognitive NeuroGenetics sample. Twin Res. Hum. Genet. 15, 442–452 (2012)
Ripke, S. et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159 (2013)
Nothnagel, M., Ellinghaus, D., Schreiber, S., Krawczak, M. & Franke, A. A comprehensive evaluation of SNP genotype imputation. Hum. Genet. 125, 163–171 (2009)
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009)
Lunter, G. & Goodson, M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21, 936–939 (2011)
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009)
Van der Auwera, G. A. et al. in Current Protocols in Bioinformatics (eds Baxevanis, A. D. et al.) vol. 11, 1–11 (Wiley, 2013)
Magocˇ, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011)
Kiełbasa, S. M., Wan, R., Sato, K., Horton, P. & Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493 (2011)
Deorowicz, S., Kokot, M., Grabowski, S. & Debudaj-Grabysz, A. KMC 2: fast and resource-frugal k-mer counting. Bioinformatics 31, 1569–1576 (2015)
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011)
Šošic´, M. & Šikic´, M. Edlib: a C/C ++ library for fast, exact sequence alignment using edit distance. Bioinformatics btw753 (2017)
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44 (D1), D81–D89 (2016)
Stucky, B. J. SeqTrace: a graphical tool for rapidly processing DNA sequencing chromatograms. J. Biomolec. Techniq. 23, 90–93 (2012)
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000)
Eberle, M. A. et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 27, 157–164 (2016)
Auer P. L. et al. Imputation of exome sequence variants into population-based samples and blood-cell-trait-associated loci in African Americans: NHLBI GO Exome Sequencing Project. Am. J. Hum. Genet. 91, 794–808 (2012)
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990)
Frith, M. C., Hamada, M. & Horton, P. Parameters for accurate genome alignment. BMC Bioinformatics 11, 80 (2010)
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open 4.0 (2013)
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013)
Oven, M., Geystelen, A., Kayser, M., Decorte, R. & Larmuseau, M. H. D. Seeing the wood for the trees: a minimal reference phylogeny for the human Y chromosome. Hum. Mutat. 35, 187–191 (2014)
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013)
Helgason, A. et al. The Y-chromosome point mutation rate in humans. Nat. Genet. 47, 453–457 (2015)
Acknowledgements
We thank the anonymous donors. The study was funded by Innovation Fund Denmark and the Novo Nordisk Foundation (grant NNF14CC0001).
Author information
Authors and Affiliations
Contributions
A.K., L. B., T.I.A.S., O.P., R.G., J.W., A.D.B., H.E., K.K., S.Br., and M.H.S. designed the study or supervised analyses. S.L., T.E., P.F.S., C.M.H., H.E., D.W., S.L.H., and P.S. provided samples. X.X., N.L., C.Y., T.H., H.C., X.G., J.S., R.X., J.R., W.Y., Y.C., and S.H. performed sequencing. T.D.A., F.L., B.P., S.R., K.B., J.M.G.I., Si.L., T.S.P., O.M., J.G., M.G., O.L., M.L.M., R.Y., C.T.H., J.V.B., L.M., J.A.S., J.B.J., M.H.S., J.M.J., L.S., C.N.S.P., T.M., P.V., R.F., Sh.L., S.H., Y.C., and S.Be. analysed data. A.S., R.F., A.E.H., E.F., P.L., A.B., M.H.S., and S.Br. provided computational infrastructure. J.A.S., L.M., S.R., S.Be., P.V., S.L., L.S., J.M.J., S.Br., and M.H.S. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks D. Church, P. de Bakker and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 The scaffold break rate and the coverage.
a, The proportion of the genome covered by the 100 largest scaffolds for the 150 assemblies. For comparison, the break rates of the assembly from Fig. 1, a published long-read assembly (PMID), and other published assemblies are shown below the distribution. b, The proportion of the 100 largest scaffolds that break when mapped against GHRC38 for the 150 assemblies. For comparison, the break rates of the assembly from Fig. 1, a published long-read assembly (BioNano–PacBio), and other published assemblies are shown below the distribution.
Extended Data Figure 2 Quality and coverage of the long-read assembly BioNano–PacBio and the haploid mole.
a, As in Fig. 1, the scaffolds that map to each chromosome, coloured by their rank in length, are shown. Regions not covered by any scaffolds in any of the 150 assemblies (thin blue line) are also seen to be problematic for this assembly. Compared with Fig. 1, many large assembly gaps in other regions are evident. b, Mapping of scaffolds for the haploid mole.
Extended Data Figure 3 Comparison of assembly statics with 150 assemblies and BioNano–PacBio assembly.
a, Comparison of the total N bases for the 150 GenomeDenmark Allpaths-LG assemblies and the BioNano–PacBio assembly. b, The average gap size distribution for 150 GenomeDenmark Allpaths-LG assemblies. Error bars, s.d. over genomes. c, The gap size distribution for the BioNano–PacBio assembly.
Extended Data Figure 4 Comparison of assembly statistics for three different methods.
a, Comparison of the scaffold N10–N90 for Danish individuals by three assemblers (Allpaths-LG, SGA, and SOAPdenovo2). b, Comparison of the scaftig N10–N90. Scaftig refers to contigs within scaffold without missing sequence. After sorting the scaffold/scaftigs in descending order on the basis of length, N10–N90 refers to the minimum length that the assembled sequences with length equal to and above that constitute 10–90% of the total scaffold size. Error bars, s.d. over all genomes.
Extended Data Figure 5 Trio transmission validation and variant size spectra.
a, Trio concordance (1 − Mendelian error rate) for the 50 trios for each variant type, where ‘Multi’ refers to multi-allelic variants and ‘Nested’ refers to variants nested within a structural variant. b, Trio concordance as a function of structural variant length for bi-allelic insertions and deletions, and conditioned on whether the variant is repetitive or not. c, Extended variant size spectrum for insertions and deletions of all called and novel insertions and deletions. d, Variant size spectrum showing the contribution of mapping-based and assembly-based callers to the set of called variant insertions and deletions.
Extended Data Figure 6 Novel GenomeDenmark variants in NA12878.
BayesTyper was used to genotype NA12878 on the basis of 50× Illumina sequencing data and a set of candidate variants was obtained by merging variant calls from GATK HaplotypeCaller, Platypus, and FreeBayes with variants from the GenomeDenmark panel and 1000 Genomes Project structural variants. a, Bar widths indicate the number of variants called for each variant class and the proportion of blue colour indicates the fraction of variants that originated from novel GenomeDenmark calls on a linear scale. b, The length distribution of the called insertion and deletion alleles is symmetrical. Note that insertions with ambiguous bases originating from inter-scaffold gaps were included although their size estimate was associated with some uncertainty. c, The proportion of variants that are novel as a function of their size. d, Trio concordance (1 − Mendelian error rate) for each variant allele class, where ‘Multi’ refers to multi-allelic variants and ‘Nested’ refers to variants nested within a structural variant. e, Trio concordance as a function of variant allele length for bi-allelic insertions and deletions.
Extended Data Figure 7 Tagability of GenomeDenmark variants.
The cumulative proportion of structural variants (y axis) in R2 linkage disequilibrium (LD) (x axis) with a single nucleotide variant. Linkage disequilibrium of structural variants is divided into (a) deletions, insertions, and complex variants on the basis of the human reference genome, into (b) small (<10 bp), medium (10–50 bp), and large (>50 bp) structural variants, and into (c) different repeat types. For each structural variant, the maximal R2 to an SNV was identified on the basis of linkage disequilibrium calculations using plink version 1.9 for all autosomes with a window size of 250 kb and 1,000 variants.
Extended Data Figure 8 GWAS tagability.
a, Proportions of exonic, intronic, and intergenic structural variants that are strongly in linkage disequilibrium with the GWAS SNPs for different functional categories of GWAS SNPs. Intergenic GWAS variants are often most strongly in linkage disequilibrium with intergenic structural variants, and intronic GWAS variants are more in linkage disequilibrium with intronic structural variants, but also relatively more often in linkage disequilibrium with exonic structural variants. A total of 954 of the structural variants are exonic and 38 of these are larger than 50 base pairs. b, Maximal linkage disequilibrium measured as the R2 correlation between a given GWAS SNP and structural variant is shown cumulatively for decreasing proportions of 28K GWAS SNPs with a P value of at most 5 × 10−8 extracted from gwasdb (http://jjwanglab.org/gwasdb). The linkage disequilibrium is generally much higher for GWAS variants than it is for all SNP variants. Differences may also be observed with respect to the type of structural variant most strongly in linkage disequilibrium with a GWAS SNP. GWAS variants in linkage disequilibrium with large structural variants are often in stronger linkage disequilibrium than smaller structural variants. A similar observation can be made for complex structural variants.
Extended Data Figure 9 Summary of the results of the imputation on clinical data.
The GOYA genotype array data set after passing the quality control contains 5,222 individuals and 514,705 SNPs. a, The number of additional variants imputed with our GenomeDenmark (GDK) call-set every 2,000 kb. These variants are imputed with the merged panel of the 1000 Genomes Project reference panels (1KG) and GenomeDenmark, but are not imputed with the 1000 Genomes Project alone. In green are the regions where the percentage of unknown bases on the reference genome is higher than 15% (>15% ‘N’). b, Median quality (R2) of the imputation per minor allele frequency, with the three reference panels. c, The number of variants imputed and filtered (information score above 0.882) selected variants, and the percentage of insertion or deletions (indels), for our new reference panel (GenomeDenmark), the 1000 Genomes Project reference panel (1000 Genomes Project), the two panels merged (GenomeDenmark plus 1000 Genomes Project), and the additional variants imputed by GenomeDenmark; see a for explanations (GenomeDenmark plus 1000 Genomes Project due to GenomeDenmark).
Extended Data Figure 10 Association mapping using the GenomeDenmark call-set.
Association of SNV and structural variants in the FTO region of the GOYA data set known to be associated with obesity. The origin of the marker is either from the 1000 Genomes Project (blue) or the GenomeDenmark (yellow) variant call-set. The GenomeDenmark SNVs are also known from the 1000 Genomes Project. Red stars show structural variation from GenomeDenmark; among these is one marker of high association close to rs9939609.
Supplementary information
Supplementary Information
This file contains information regarding the de novo assembly, the variant calling and genotyping, protein truncating and genotyping, PTV, de novo variation, the extended MHC region and Y chromosome, genome-graph representations, cohort selection and raw data. (PDF 3196 kb)
Supplementary Table 1
This file contains detailed statistics for AllPathsLG denovo assemblies of 150 individual genomes. (XLSX 122 kb)
Supplementary Table 2
This file contains comparison of AllPathsLG, SGA and Soapdenovo de novo assemblies for each of 150 individual genomes. (XLSX 127 kb)
Supplementary Table 3
This file contains details on HLA validation data. (XLSX 14 kb)
Supplementary Table 4
This file contains details on Y chromosome validation data. (XLSX 11 kb)
Supplementary Table 5
This file contains details on production sequencing data. (XLSX 103 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maretty, L., Jensen, J., Petersen, B. et al. Sequencing and de novo assembly of 150 genomes from Denmark as a population reference. Nature 548, 87–91 (2017). https://doi.org/10.1038/nature23264
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature23264
This article is cited by
-
Rare coding variants in NOX4 link high ROS levels to psoriatic arthritis mutilans
EMBO Molecular Medicine (2024)
-
A review of the pangenome: how it affects our understanding of genomic variation, selection and breeding in domestic animals?
Journal of Animal Science and Biotechnology (2023)
-
Evolution of the germline mutation rate across vertebrates
Nature (2023)
-
SVDSS: structural variation discovery in hard-to-call genomic regions using sample-specific strings from accurate long reads
Nature Methods (2023)
-
Haplotype-aware pantranscriptome analyses using spliced pangenome graphs
Nature Methods (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
