
Abstract
D-type cyclins (D1, D2 and D3) and their associated cyclin-dependent kinases (CDK4 and CDK6) are components of the core cell cycle machinery that drives cell proliferation1,2. Inhibitors of CDK4 and CDK6 are currently being tested in clinical trials for patients with several cancer types, with promising results2. Here, using human cancer cells and patient-derived xenografts in mice, we show that the cyclin D3–CDK6 kinase phosphorylates and inhibits the catalytic activity of two key enzymes in the glycolytic pathway, 6-phosphofructokinase and pyruvate kinase M2. This re-directs the glycolytic intermediates into the pentose phosphate (PPP) and serine pathways. Inhibition of cyclin D3–CDK6 in tumour cells reduces flow through the PPP and serine pathways, thereby depleting the antioxidants NADPH and glutathione. This, in turn, increases the levels of reactive oxygen species and causes apoptosis of tumour cells. The pro-survival function of cyclin D-associated kinase operates in tumours expressing high levels of cyclin D3–CDK6 complexes. We propose that measuring the levels of cyclin D3–CDK6 in human cancers might help to identify tumour subsets that undergo cell death and tumour regression upon inhibition of CDK4 and CDK6. Cyclin D3–CDK6, through its ability to link cell cycle and cell metabolism, represents a particularly powerful oncoprotein that affects cancer cells at several levels, and this property can be exploited for anti-cancer therapy.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Malumbres, M. Cyclin-dependent kinases. Genome Biol. 15, 122 (2014)
Sherr, C. J., Beach, D. & Shapiro, G. I. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 6, 353–367 (2016)
Deshpande, A., Sicinski, P. & Hinds, P. W. Cyclins and CDKs in development and cancer: a perspective. Oncogene 24, 2909–2915 (2005)
Dean, J. L., Thangavel, C., McClendon, A. K., Reed, C. A. & Knudsen, E. S. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 29, 4018–4032 (2010)
Puyol, M. et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 18, 63–73 (2010)
Dean, J. L. et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 11, 2756–2761 (2012)
Choi, Y. J. et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell 22, 438–451 (2012)
Sawai, C. M. et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 22, 452–465 (2012)
Cairns, R. A., Harris, I. S. & Mak, T. W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 (2011)
Christofk, H. R. et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233 (2008)
Tong, X., Zhao, F. & Thompson, C. B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 19, 32–37 (2009)
Banaszak, K. et al. The crystal structures of eukaryotic phosphofructokinases from baker’s yeast and rabbit skeletal muscle. J. Mol. Biol. 407, 284–297 (2011)
Brüser, A., Kirchberger, J., Kloos, M., Sträter, N. & Schöneberg, T. Functional linkage of adenine nucleotide binding sites in mammalian muscle 6-phosphofructokinase. J. Biol. Chem. 287, 17546–17553 (2012)
Schöneberg, T., Kloos, M., Brüser, A., Kirchberger, J. & Sträter, N. Structure and allosteric regulation of eukaryotic 6-phosphofructokinases. Biol. Chem. 394, 977–993 (2013)
Mazurek, S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 43, 969–980 (2011)
Anastasiou, D. et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334, 1278–1283 (2011)
Boada, J. et al. Cells overexpressing fructose-2,6-bisphosphatase showed enhanced pentose phosphate pathway flux and resistance to oxidative stress. FEBS Lett. 480, 261–264 (2000)
Chaneton, B. et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491, 458–462 (2012)
Yi, W. et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337, 975–980 (2012)
Fan, J. et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302 (2014)
Phong, W. Y. et al. Characterization of phosphofructokinase activity in Mycobacterium tuberculosis reveals that a functional glycolytic carbon flow is necessary to limit the accumulation of toxic metabolic intermediates under hypoxia. PLoS One 8, e56037 (2013)
Kotlarz, D. & Buc, H. Phosphofructokinases from Escherichia coli. Methods Enzymol. 90, 60–70 (1982)
Cetica, P., Pintos, L., Dalvit, G. & Beconi, M. Activity of key enzymes involved in glucose and triglyceride catabolism during bovine oocyte maturation in vitro. Reproduction 124, 675–681 (2002)
Divita, G., Goody, R. S., Gautheron, D. C. & Di Pietro, A. Structural mapping of catalytic site with respect to alpha-subunit and noncatalytic site in yeast mitochondrial F1-ATPase using fluorescence resonance energy transfer. J. Biol. Chem. 268, 13178–13186 (1993)
Vertommen, D. et al. The ATP-binding site in the 2-kinase domain of liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Study of the role of Lys-54 and Thr-55 by site-directed mutagenesis. J. Biol. Chem. 271, 17875–17880 (1996)
Nicolay, B. N. et al. Loss of RBF1 changes glutamine catabolism. Genes Dev. 27, 182–196 (2013)
Antoniewicz, M. R., Kelleher, J. K. & Stephanopoulos, G. Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab. Eng. 8, 324–337 (2006)
Antoniewicz, M. R., Kelleher, J. K. & Stephanopoulos, G. Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab. Eng. 9, 68–86 (2007)
Yoo, H., Antoniewicz, M. R., Stephanopoulos, G. & Kelleher, J. K. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J. Biol. Chem. 283, 20621–20627 (2008)
Young, J. D., Walther, J. L., Antoniewicz, M. R., Yoo, H. & Stephanopoulos, G. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng. 99, 686–699 (2008)
Marin-Valencia, I. et al. Glucose metabolism via the pentose phosphate pathway, glycolysis and Krebs cycle in an orthotopic mouse model of human brain tumors. NMR Biomed. 25, 1177–1186 (2012)
Huttlin, E. L. et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010)
Eng, J. K., McCormack, A. L. & Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 (1994)
Elias, J. E. & Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007)
Beausoleil, S. A., Villén, J., Gerber, S. A., Rush, J. & Gygi, S. P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 (2006)
Gao, H. et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 21, 1318–1325 (2015)
Acknowledgements
Supported by R01 CA083688, R01 CA202634, P01 CA080111 (P.S.), R01 CA163698 (N.J.D.) and F32 CA165856 (B.N.N.). N.J.D. is a James and Shirley Curvey MGH Research Scholar. X.G. was supported by an NIH post-doc training grant (T32CA009361) and NIH grant P50 CA090381-14 (DF/HCC SPORE in Prostate Cancer). J.M.S. was supported by a Mobilnos´c´ Plus fellowship. We thank M. Eck, J. Daly, P. Hydbring, T. Otto, W. Michowski and I. Harris for help.
Author information
Authors and Affiliations
Contributions
H.W. and P.S. designed the study. H.W. performed all experiments with the help of collaborators as follows. B.N.N. and N.J.D. performed and interpreted isotopic enrichment analyses. J.M.C. and S.P.G. contributed mass spectrometric and biocomputational analyses. X.G. helped with viral transductions and kinase assays. Y.G. helped with T-ALL xenografts and kinase assays. H.R. helped with analyses of cyclin levels and tissue culture. J.M.S. helped with design and construction of expression vectors. T.M.R. helped with supervision. H.G., G.Y. and J.A.W. contributed ribociclib xenograft studies. M.A.K. contributed some isotopic-enrichment analyses. E.S. carried out pathological analyses. U.G. and W.N.H. isolated human T cells. K.P. helped with breast cancer studies. H.W. and P.S. wrote the paper. P.S. directed the study.
Corresponding author
Ethics declarations
Competing interests
P.S., T.M.R. and K.P. are consultants and recipients of research grants from Novartis. H.G., G.Y. and J.A.W. are employees of Novartis Institutes for BioMedical Research.
Additional information
Reviewer Information Nature thanks J. Bartek, H. Christofk and A. Schulze for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Analyses of human T-ALL cells.
a, b, Quantification of D-type cyclin levels (a), CDK4 and CDK6 (b) in T-ALL cell lines. c, CDK6 was immunoprecipitated and immunoblots probed with the indicated antibodies. d, T-ALL cell lines were transduced with viruses encoding shRNA against cyclin D3, CDK6 or control shRNA (–), and apoptosis was gauged by annexin V staining and FACS. e, Immunoblotting from d. f–h, The goal was to determine whether inhibition of CDK4/6 causes apoptosis of normal human T lymphocytes. T lymphocytes from two donors (#1, #2) were stimulated with interleukin 2 (IL-2) and analysed by immunoblotting (f), or cultured in the presence or absence of palbociclib (PALBO), and analysed for apoptosis as in d (g), or cultured as in g, pulsed with BrdU, stained with an anti-BrdU antibody and propidium iodide, and analysed for cell cycle distribution (h). i, T-ALL cell lines were transduced with viruses encoding anti-RB1 or control shRNA (–). Cells were cultured in the presence or absence of palbociclib, and apoptosis was quantified as in d. j, Cells were treated as in i, and apoptosis was assessed by immunoblotting with anti-cleaved PARP antibody. k, T-ALL cell lines were transduced with viruses encoding shRNAs against RB1, RBL1 and RBL2 or control shRNA (–). Cells were cultured in the presence or absence of palbociclib, and apoptosis quantified as in d. l, Immunoblot analysis from k. m, Quantification of expression of PFK1 isoforms in T-ALL cell lines. n, Analysis of expression of PKM1 and PKM2 in T-ALL cell lines and in lung cancer A549 and embryonic kidney 293T cells. o, Quantification of expression of PKM2 in T-ALL cell lines. p, q, Interaction of PFKP (p) and PKM2 (q) with CDK6 and cyclin D3 in T-ALL cell lines. HC, Ponceau staining for immunoglobulin heavy chains. In a–c, i, j, m–q, identical results were obtained with RPMI8402 cells, which are not shown (lines spliced out) owing to their inclusion in the list of potentially misidentified cell lines. d, g–i, k, n = 3, biological replicates. Bars, mean values; error bars, s.d. **P < 0.01, ***P < 0.001 (t-test). a, b, Representative experiments out of two; c, e, f, j, l–q, out of three independent experiments. Supplementary Fig. 1 shows gel source data.
Extended Data Figure 2 Phosphorylation of PFK1 and PKM2 by cyclin D3–CDK6.
a, In vitro kinase reactions. Pon S, Ponceau S staining of membranes. Arrows point to PFK1 or PKM2 proteins; star, phosphorylation of CDK4. b, In vitro kinase reactions (controls for analyses shown in a). c, In vitro kinase reactions to test whether cyclin D3–CDK6 can phosphorylate PKM1, PKR or PKL isoforms of pyruvate kinase. Stars denote phosphorylation of cyclin D3 and CDK6. d, Cyclin D3–CDK6-dependent phosphoresidues, identified in our mass spectrometric analyses. In the case of PFKM we were unable to distinguish between phosphorylation of S667 and T669. e, Decreased phosphorylation of endogenous PFKP and PKM2 upon palbociclib treatment. T-ALL cell lines were cultured in the presence or absence of palbociclib. PFKP or PKM2 were immunoprecipitated and immunoblots probed with an anti-phospho Ser-Pro/Thr-Pro antibody (Phos-SP/TP) to detect phosphorylated PFKP, or with an antibody against serine 37-phosphorylated PKM2 (Phos-S37-PKM2), or with anti-PFKP and -PKM2 antibodies. Whole-cell extracts (WCE) were also immunoblotted. Quantification of band intensities is shown in Fig. 1c. Identical results were obtained with RPMI8402 cells (lines spliced out). f, Decreased phosphorylation of endogenous PFKP and PKM2 upon depletion of cyclin D3. T-ALL cell lines were transduced with viruses encoding shRNA against cyclin D3, or control shRNA (–). PFKP or PKM2 phosphorylation was assessed as in e. Arrows point to PKM2; stars, immunoglobulin heavy chain. g, Quantification of PFKP and PKM2 phosphorylation from f. h, Immunoblot analysis from f. i, Decreased phosphorylation of PFKP and PKM2 upon depletion of CDK6. T-ALL cell lines were transduced with viruses encoding shRNA against CDK6, or control shRNA (–). PFKP or PKM2 phosphorylation was assessed as in e and f. j, Quantification of PFKP and PKM2 phosphorylation from i. k, Immunoblot analysis from i. g, j, Quantification of experiments shown in f and i, respectively (out of three biological replicates). Bars, mean values; error bars, s.d. **P < 0.01,***P < 0.001 (t-test). a, b, e, f, h, i, k, Representative experiments out of three; c, out of five independent experiments. Supplementary Fig. 1 shows gel source data.
Extended Data Figure 3 Cyclin D3–CDK6 inhibits PFKP and PKM2.
a, b, Purified recombinant wild-type or mutant PFKP (a) or PKM2 (b) was preincubated with (+) or without (–) cyclin D3–CDK6, and PFKP or PKM2 activity was assayed. c, d, Cells were transduced with viruses encoding shRNA against cyclin D3 or CDK6 or control shRNA (–), and the activity of PFKP (c) or PKM2 (d) was assayed. e, Purified recombinant wild-type or mutant PFKP was preincubated as in a, and ADP binding was assayed using 100 μM ADP and 18 pmol PFKP. f, g, Binding of PFKP to ADP was assayed using 100 μM ADP (f) or 22.3 pmol PFKP (g). h, Flag-tagged wild-type or mutant PFKP was expressed in MCF7 cells. Whole-cell lysates were resolved on a non-denaturing gel and immunoblots probed with an anti-Flag antibody. The amount of protein extract loaded onto gels was adjusted so that each sample contained roughly the same amount of PFKP tetramers. Lower panels, whole-cell lysates were resolved on a denaturing SDS–PAGE gel. i, Quantification from h. j, Flag-tagged PFKP was expressed in MOLT4 cells. Whole-cell lysates were filtered using Disposable Ultrafiltration Units with molecular mass 200 kDa cutoff. The filtrates (<200 kDa fraction) were then analysed, along with whole-cell extracts (WCE) before filtration, by immunoblotting with an anti-Flag antibody. LRP6 protein (molecular mass 180 kDa, passes the filter), BRCA1 (208 kDa, not passing the filter). k, Quantification from j. l, Cell lines were cultured in the presence or absence of palbociclib. Whole-cell lysates were filtered as in j. m, Quantification from l. n, Flag-tagged PKM2 was expressed in EBC1 (lanes 1, 2) and HUCCT1 cells (lanes 3, 4). Whole-cell lysates were resolved on a non-denaturing gel and immunoblots probed with an anti-Flag antibody. Lower panel, whole-cell lysates were resolved on a denaturing SDS–PAGE gel. o, Quantification from n. a–g, i, k, m, o, Bars, mean values; error bars, s.d. *P < 0.05, **P < 0.01, ***P < 0.001 (t-test). a–g, n = 3 independent experiments. i, k, m, o, Quantification of experiments in h, j, l, n respectively (out of three independent experiments). h, Representative experiment out of five; j, l, n, out of three. Supplementary Fig. 1 shows gel source data.
Extended Data Figure 4 Metabolic changes in T-ALL cells upon inhibition of cyclin D3–CDK6.
a, b, Flow of glucose-derived carbon into the PPP following cyclin D3–CDK6 inhibition in T-ALL KOPTK1 (a) and MOLT4 (b) cells. Cells were fed with isotopically labelled [1,2-13C]glucose in the presence or absence of palbociclib, and the flow of 13C was quantified using isotopic enrichment analysis and mass spectrometry. c, d, Flow of glucose-derived carbon into the serine pathway following cyclin D3–CDK6 inhibition in T-ALL KOPTK1 (c) and MOLT4 (d) cells. e–g, Cells were transduced with viruses encoding shRNA against cyclin D3 or CDK6, or control shRNA (–), and metabolite levels were measured. h, i, Cells were cultured in the presence or absence of palbociclib and NAC (h), or palbociclib and MnTMPyP (i), and ROS levels were measured. j, Cells were cultured in the presence or absence of palbociclib and MnTMPyP, and apoptosis was gauged using a cleaved caspase 3/7 assay. a–j, n = 3, biological replicates. Bars, mean values; error bars, s.d. *P < 0.05, **P < 0.01, ***P < 0.001 (t-test).
Extended Data Figure 5 Functional consequences of PFKP and PKM2 phosphorylation by cyclin D3–CDK6.
a, Analysis of PFKP and PKM2 levels in T-ALL KOPTK1 and MOLT4 cells that were left untreated (non), or engineered to express control shRNA (shcon), or anti-PFKP and anti-PKM2 shRNAs (shPF + shPK), or anti-PFKP and anti-PKM2 shRNAs together with ectopically expressed wild-type PFKP and PKM2 (shPF + shPK + WT + WT), or anti-PFKP and anti-PKM2 shRNAs together with ectopically expressed PFKP-S679E and PKM2-S37E phosphomimicking mutants (shPF + shPK + S679E + S37E). b–e, Cells engineered as in a to express wild-type PFKP and PKM2 (KOPTK1-WT, MOLT4-WT), or PFKP-S679E and PKM2-S37E mutants (KOPTK1-EE, MOLT4-EE), were cultured in the presence or absence of palbociclib, and the activities of PFKP and PKM2 were measured. f, KOPTK1-EE and MOLT4-EE cells were cultured in the presence or absence of palbociclib. Whole-cell lysates were filtered and analysed as in Extended Data Fig. 3j. g, Quantification from f. h–j, Flow of glucose-derived carbon into the PPP (h, i) and the serine pathway (j) following cyclin D3–CDK6 inhibition in KOPTK1-WT and KOPTK1-EE or MOLT4-WT and MOLT4-EE cells, analysed as in Extended Data Fig. 4a, b. k–p, KOPTK1-WT and KOPTK1-EE or MOLT4-WT and MOLT4-EE cells were cultured in the presence or absence of palbociclib and metabolite levels were determined. q, Apoptosis was quantified by cleaved caspase 3/7 assay. r, s, Apoptosis analysed by annexin V staining followed by FACS. t, Quantification from s. u, KOPTK1-WT and KOPTK1-EE cells were transduced with viruses encoding shRNA against CDK6, or control shRNA (–), and apoptosis was analysed as in r. v, Immunoblot analysis from u and Fig. 2h. w, x, Cells were treated as indicated and apoptosis quantified as in r. y, z, Fraction of cells in the indicated cell cycle phases. b–e, g–q, t, u, w, x, Bars, mean values; error bars, s.d. *P < 0.05, **P < 0.01, ***P < 0.001 (t-test). b–e, h–q, u, w, x, n = 3 biological replicates. g, t, Quantification of the experiments shown in f, s, respectively (out of three independent experiments). a, f, v, Representative experiments (out of three independent experiments). r, y, z, n = 3 independent experiments. Supplementary Fig. 1 shows gel source data.
Extended Data Figure 6 Analyses of KOPTK1 and MOLT4 T-ALL cells expressing phosphomutant PFKP or PKM2.
a–i, To gauge the contribution of PFKP versus PKM2 phosphorylation to the observed phenotypes, we engineered single mutant T-ALL cells expressing phosphomimicking PFKP-S679E in place of wild-type PFKP (along with wild-type PKM2), and cells expressing phosphomimicking PKM2-S37E in place of wild-type PKM2, (along with wild-type PFKP). a, Immunoblot analysis of PFKP and PKM2 levels in T-ALL KOPTK1 and MOLT4 cells engineered to stably express control shRNA (shcon), or anti-PKM2 shRNA together with ectopically expressed wild-type PKM2 (shPK + WT), or anti-PKM2 shRNAs together with PKM2-S37E (shPK + S37E), or anti-PFKP shRNA plus ectopically expressed wild-type PFKP (shPF + WT), or anti-PFKP shRNAs plus PFKP-S679E (shPF + S679E). b–i, Analysis of single mutant KOPTK1 and MOLT4 cells engineered as in a to express wild-type PFKP and PKM2 (WT), or PKM2-S37E (PK-S37E), or PFKP-S679E (PF-S679E). Cells were cultured in the presence or absence of palbociclib, and the indicated parameters were measured. Note that palbociclib treatment of single mutant cells reduced PPP and serine pathway flows and GSH levels, increased ROS and triggered apoptosis; the effects were generally milder than those in cells expressing wild-type PFKP and PKM2. j–n, Analyses of KOPTK1 and MOLT4 T-ALL cells expressing doxycycline-inducible phospho-inactivating PFKP-S679A and PKM2-S37A mutants. j, Immunoblot analysis of PKM2 and PFKP levels in T-ALL MOLT4 cells engineered to stably express control shRNA (shcon), or anti-PFKP and anti-PKM2 shRNAs together with doxycycline-inducible wild-type PFKP and PKM2 (shPF + shPK + WT + WT), or anti-PFKP and anti-PKM2 shRNAs together with doxycycline-inducible PFKP-S679A and PKM2-S37A (shPF + shPK + S679A + S37A). Cells were cultured in the absence or presence of doxycycline (DOX). k–n, MOLT4 cells engineered as in j to express doxycycline-inducible wild-type PFKP and PKM2 (WT), or PFKP-S679A and PKM2-S37A (AA) were cultured in the presence or absence of doxycycline and the indicated parameters were measured. b–i, k–n, n = 3 biological replicates. Bars, mean values; error bars, s.d. *P < 0.05, **P < 0.01, ***P < 0.001 (t-test). a, j, Representative experiment (out of two independent experiments). Supplementary Fig. 1 shows gel source data.
Extended Data Figure 7 Inhibition of PPP and serine pathway in T-ALL cells.
a–d, KOPTK1 cells were transduced with vectors encoding shRNA against phosphoserine aminotransferase, the key enzyme in the serine pathway (shPSAT +), or control shRNA (–), and cultured in the presence (+) or absence (–) of dehydroepiandrosterone (DHEA, an inhibitor of the PPP), and the levels of NADPH (a), reduced glutathione (GSH, b), ROS (c), and apoptosis (d, annexin V staining followed by FACS) were determined. e–h, Similar analysis of MOLT4 cells. i–l, Similar analysis of DND41 cells. m, Immunoblot analysis to gauge the efficiency of PSAT1 knockdown. Tubulin served as a loading control. n–p, The indicated T-ALL cell lines were treated with vehicle (con) or with PPP inhibitors 6-AN or DHEA, and the levels of NADPH (n) and ROS (o) were assayed. Apoptosis was quantified by annexin V staining followed by FACS (p). a–l, n–p, n = 3 biological replicates. Bars, mean values; error bars, s.d. *P < 0.05, **P < 0.01, ***P < 0.001 (t-test). m, Representative experiment (out of three independent experiments). Supplementary Fig. 1 shows gel source data.
Extended Data Figure 8 Analyses of human breast cancer cells.
a, Apoptosis levels in human breast cancer cell lines cultured in the presence or absence of palbociclib (cleaved caspase 3/7 assay). b, Protein levels in breast cancer and T-ALL cell lines, assessed by immunoblotting of whole-cell lysates. c, Breast cancer cell lines were cultured in the presence or absence of palbociclib; PFKP and PKM2 phosphorylation was assessed as in Extended Data Fig. 2e. Arrow points to PKM2; star, immunoglobulin heavy chain. Right, immunoblotting of whole-cell extracts (WCE). d, Quantification from c. e, Breast cancer cell lines were cultured in the presence or absence of palbociclib. Whole-cell lysates were filtered and analysed as in Extended Data Fig. 3j. f, Quantification from e. g–k, Breast cancer cell lines were cultured in the presence or absence of palbociclib, and the indicated parameters were assessed. l, Absolute levels of ROS in human T-ALL and breast cancer cell lines. Identical results were obtained in T-ALL RPMI8402 cells. Note that the absolute levels of ROS in breast cancer cell lines are overall higher than those in T-ALL cells, which may render breast cancer cells less sensitive to modest increases in ROS levels observed in these cells upon CDK4/6 inhibition (see k). m, n, Breast cancer cells were transduced with vectors encoding shRNA against phosphoserine aminotransferase (PSAT), the key enzyme in the serine pathway or control shRNA (–), and cultured in the presence or absence of 6-AN, and the indicated parameters were measured. o, Immunoblot analysis from n. p, ROS levels in breast cancer cells treated with palbociclib alone, or together with a low concentration of 6-AN. Note that palbociclib treatment caused only a small increase in ROS, which was enhanced by 6-AN. q, Cells were treated with palbociclib together with low concentrations of 6-AN, and apoptosis quantified by annexin V/FACS. Note the synergistic effect of palbociclib and 6-AN in inducing apoptosis. a, d, f–n, p, q, Bars, mean values; error bars, s.d. *P < 0.05, **P < 0.01, ***P < 0.001 (t-test). a, g–n, p, q, n = 3, biological replicates. d, f, Quantification of c, e, respectively (out of three biological replicates). b, c, e, o, Representative experiments (out of three independent experiments). Supplementary Fig. 1 shows gel source data.
Extended Data Figure 9 Apoptosis of cyclin D3/CDK6-high tumour cell lines upon inhibition of CDK4 and CDK6.
a, Transcript levels (log2 Robust Multi-Array Average values) according to CCLE (https://portals.broadinstitute.org/ccle/home). Left, CDK6-high; right, CDK6-low cell lines. b, Immunoblot analysis of ‘top 20’ cell lines. T98G and SW620 cells (arrows) express lower CDK6 and/or cyclin D3 protein levels. c, d, Apoptosis of D3/CDK6-high cell lines upon palbociclib treatment (c), or cyclin D3 or CDK6 knockdown (d). e, Immunoblot analysis from d. f, Analysis of T98G and SW620 cells. g, h Immunoblot analysis and apoptosis of D3/CDK6-low cell lines. i, D3/CDK6-high cells analysed as in Extended Data Fig. 2e. Arrows point to PKM2; stars, immunoglobulin heavy chain. Quantification is shown in Fig. 4a, b. j, D3/CDK6-high cells were transduced with anti-CDK6 or control shRNA (–); PFKP and PKM2 phosphorylation was assessed as above. k, Quantification from j. l–n, Analysis of D3/CDK6-low cells. o, D3/CDK6-high HUCCT1 cells expressing wild-type PFKP and PKM2 (HUCCT1-WT) or PFKP-S679E and PKM2-S37E (HUCCT1-EE) were cultured in the presence or absence of palbociclib. p–r, To gauge the contribution of PFKP versus PKM2 phosphorylation to the observed phenotypes, we engineered single mutant D3/CDK6-high EBC1 and HUCCT1 cells expressing PFKP-S679E in place of wild-type PFKP (along with wild-type PKM2), and cells expressing PKM2-S37E in place of wild-type PKM2 (along with wild-type PFKP). p, Immunoblot analysis of PKM2 and PFKP levels in EBC1 and HUCCT1 cells expressing control shRNA (shcon), anti-PKM2 shRNA (shPK), anti-PKM2 shRNA plus ectopically expressed wild-type PKM2 (shPK + WT), anti-PKM2 shRNA plus PKM2-S37E (shPK + S37E), anti-PFKP shRNA plus wild-type PFKP (shPF + WT), or anti-PFKP shRNA plus PFKP-S679E (shPF + S679E). q, r, Analysis of cells engineered in p to express wild-type PKM2 and PFKP (WT), or mutants (PK-S37E or PF-S679E). Single mutant cells underwent apoptosis upon palbociclib treatment; the effect was less pronounced than in cells expressing wild-type PFKP and PKM2. c, d, f, h, k, l–o, q, r, Bars, mean values; error bars, s.d. *P < 0.05, **P < 0.01, ***P < 0.001 (t-test). c, d, f, h, l–o, q, r, n = 3, biological replicates. k, Quantification from j (out of three independent experiments). b, e, g, i, j, p, Representative experiments (out of three independent experiments). Supplementary Fig. 1 shows gel source data.
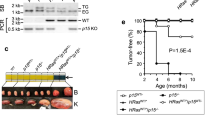
Extended Data Figure 10 Analyses of human melanoma xenografts.
Patient-derived melanomas were implanted into immunocompromised mice and the recipients treated with ribociclib or vehicle. a, b, Growth curves of 20 tumours that responded to ribociclib with reduced tumour growth but no regression (a, ribociclib-treated; b, vehicle-treated tumours). c–e, Growth curves of three tumours that underwent long-term regression upon ribociclib treatment. f, CDK6 and cyclin D3 protein levels in primary, patient-derived melanomas (before implantation into mice). Three tumours that, upon implantation into mice, underwent long-term regression in response to ribociclib are marked ‘Tumour regression’. g, Analysis of the levels of cyclin D3–CDK6 complexes in tumours shown in f. Tumour lysates from primary melanomas were immunoprecipitated with an anti-CDK6 antibody and the immunoblots were probed with the indicated antibodies. HC, Ponceau staining for immunoglobulin heavy chains. h, CDK6 and cyclin D3 protein levels in primary, patient-derived melanomas. Tumours shown here expressed low or moderate CDK6 and cyclin D3 levels, and none underwent regression upon ribociclib treatment of recipient mice (see a, b). For comparison, CDK6 and cyclin D3 levels in a melanoma that underwent long-term regression upon palbociclib treatment of recipient mice (Tumour regression) and in T-ALL KOPTK1 cells are shown. i, Analysis of three tumours (1906, 3483, 4339) that expressed high cyclin D3 and CDK6 levels (see f, g), and underwent long-term regression upon ribociclib treatment (see c–e). In this experiment, mice bearing xenografts of melanomas (n = 3 recipient mice per tumour/treatment, marked 1–3) were treated with ribociclib or vehicle. Tumours were removed 4 h after the last dose, and PFKP and PKM2 phosphorylation analysed as in Extended Data Fig. 2e. Arrows point to PKM2; stars, immunoglobulin heavy chain. Tumour lysates were also immunoblotted. Quantification is shown in Fig. 5a, b (see D3/CDK6 high). j, Similar analysis as in i of three tumours (1655, 3746, 4644) that expressed relatively low levels of cyclin D3 and/or CDK6(see f–h) and did not undergo regression upon ribociclib treatment (see a, b). Quantification is shown in Fig. 5a, b (see D3/CDK6 low). a–e, One set of experiments. f–j, Representative experiments (out of three independent experiments). Supplementary Fig. 1 shows gel source data. Source Data for Extended Data Fig. 10 shows PDX tumour growth.
Supplementary information
Supplementary Figure
This file contains the uncropped blots. (PDF 4038 kb)
Supplementary Table 1
This tablecontains affinity purification tandem mass spectrometry analysis of CDK6 interactome in T-ALL cells. (XLSX 881 kb)
Supplementary Table 2
This table contains gene set enrichment analysis of common CDK6 interactors. (XLSX 14 kb)
Supplementary Table 3
This table contains CDK6-associated glycolytic enzymes that contain potential CDK phosphorylation residues. (XLSX 44 kb)
Supplementary Table 4
This table contains LC-MS/MS analysis of cyclin D3-CDK6 dependent phosphorylation sites on PKM2 and PFKP. (XLSX 11 kb)
Rights and permissions
About this article

Cite this article
Wang, H., Nicolay, B., Chick, J. et al. The metabolic function of cyclin D3–CDK6 kinase in cancer cell survival. Nature 546, 426–430 (2017). https://doi.org/10.1038/nature22797
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature22797
This article is cited by
-
Cone photoreceptor phosphodiesterase PDE6H inhibition regulates cancer cell growth and metabolism, replicating the dark retina response
Cancer & Metabolism (2024)
-
Hijacking of nucleotide biosynthesis and deamidation-mediated glycolysis by an oncogenic herpesvirus
Nature Communications (2024)
-
Cerium oxide nanoparticles-carrying human umbilical cord mesenchymal stem cells counteract oxidative damage and facilitate tendon regeneration
Journal of Nanobiotechnology (2023)
-
Inhibition of USP10 induces myeloma cell apoptosis by promoting cyclin D3 degradation
Acta Pharmacologica Sinica (2023)
-
The RNA-binding protein LRPPRC promotes resistance to CDK4/6 inhibition in lung cancer
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
