
Abstract
Mitochondrial calcium (mCa2+) has a central role in both metabolic regulation and cell death signalling, however its role in homeostatic function and disease is controversial1. Slc8b1 encodes the mitochondrial Na+/Ca2+ exchanger (NCLX), which is proposed to be the primary mechanism for mCa2+ extrusion in excitable cells2,3. Here we show that tamoxifen-induced deletion of Slc8b1 in adult mouse hearts causes sudden death, with less than 13% of affected mice surviving after 14 days. Lethality correlated with severe myocardial dysfunction and fulminant heart failure. Mechanistically, cardiac pathology was attributed to mCa2+ overload driving increased generation of superoxide and necrotic cell death, which was rescued by genetic inhibition of mitochondrial permeability transition pore activation. Corroborating these findings, overexpression of NCLX in the mouse heart by conditional transgenesis had the beneficial effect of augmenting mCa2+ clearance, preventing permeability transition and protecting against ischaemia-induced cardiomyocyte necrosis and heart failure. These results demonstrate the essential nature of mCa2+ efflux in cellular function and suggest that augmenting mCa2+ efflux may be a viable therapeutic strategy in disease.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Murphy, E. et al. Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 449, 384–385 (2014)
Palty, R. et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl Acad. Sci. USA 107, 436–441 (2010)
Sekler, I. Standing of giants shoulders the story of the mitochondrial Na+Ca2+ exchanger. Biochem. Biophys. Res. Commun. 460, 50–52 (2015)
Antony, A. N. et al. MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat. Commun. 7, 10955 (2016)
Mallilankaraman, K. et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151, 630–644 (2012)
Mallilankaraman, K. et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336–1343 (2012)
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I. & Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 (2011)
Baughman, J. M. et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 (2011)
Perocchi, F. et al. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296 (2010)
Sancak, Y. et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382 (2013)
Bers, D. M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49 (2008)
Maack, C. & O’Rourke, B. Excitation–contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 102, 369–392 (2007)
Luongo, T. S. et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Reports 12, 23–34 (2015)
Pan, X. et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472 (2013)
Kwong, J. Q. et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Reports 12, 15–22 (2015)
Palty, R. & Sekler, I. The mitochondrial Na+/Ca2+ exchanger. Cell Calcium 52, 9–15 (2012)
Kostic, M. et al. PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Reports 13, 376–386 (2015)
Skarnes, W. C. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 (2011)
Sohal, D. S. et al. Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 89, 20–25 (2001)
Davis, J., Maillet, M., Miano, J. M. & Molkentin, J. D. Lost in transgenesis: a user’s guide for genetically manipulating the mouse in cardiac research. Circ. Res. 111, 761–777 (2012)
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M. W. & Sheu, S. S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833 (2004)
Baines, C. P. et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 (2005)
Oka, T. et al. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ. Res. 98, 837–845 (2006)
Boyman, L. et al. Calcium movement in cardiac mitochondria. Biophys. J. 107, 1289–1301 (2014)
Bers, D. M. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 21, 380–387 (2006)
Kohlhaas, M. et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121, 1606–1613 (2010)
Liu, T. & O’Rourke, B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ. Res. 103, 279–288 (2008)
Nakayama, H. et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Invest. 117, 2431–2444 (2007)
Holmström, K. M. et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell. Cardiol. 85, 178–182 (2015)
Williams, G. S., Boyman, L. & Lederer, W. J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell. Cardiol. 78, 35–45 (2015)
Reynolds, E. S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 17, 208–212 (1963)
Zhang, X. et al. Persistent increases in Ca2+ influx through Cav1.2 shortens action potential and causes Ca2+ overload-induced afterdepolarizations and arrhythmias. Basic Res. Cardiol. 111, 4 (2016)
Frezza, C., Cipolat, S. & Scorrano, L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protocols 2, 287–295 (2007)
Hamer, P. W., McGeachie, J. M., Davies, M. J. & Grounds, M. D. Evans Blue Dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J. Anat. 200, 69–79 (2002)
Dipla, K., Mattiello, J. A., Jeevanandam, V., Houser, S. R. & Margulies, K. B. Myocyte recovery after mechanical circulatory support in humans with end-stage heart failure. Circulation 97, 2316–2322 (1998)
Gao, E. et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 107, 1445–1453 (2010)
Acknowledgements
We thank T. Tierney, N. Shah, and P. Kelly for technical assistance in the Elrod Laboratory. We thank S. Modla for her expertise in TEM sample processing (Delaware Biotechnology Institute). This study was supported by grants to J.W.E. from the NIH (R01 HL123966, P01 DA037830 sub-8614) and AHA (14SDG18910041) and (15PRE25080299 to T.S.L.), (16PRE31030038 to A.A.L.), and (17PRE33460423 to J.P.L.).
Author information
Authors and Affiliations
Contributions
J.W.E. and T.S.L. contributed to study design, data analysis and writing the paper; T.S.L. was involved with all assays and data collection; J.P.L. performed western blots and NADH assays; P.G. performed ACM iCa2+ transient analysis; M.N. performed myocardial infarction data acquisition; A.A.L. assisted with the ischaemia reperfusion study and mitochondrial membrane potential assays; S.S. and M.M. assisted with efflux analysis in permeabilized cells; A.C.C. performed qPCR assays; D.K. performed histology; E.G. performed ischaemia reperfusion and myocardial infarction surgeries; J.H.v.B. and J.D.M. aided mutant mouse generation; E.J.T. supplied human heart samples; X.C. performed radiotelemeter implantation and assisted with electrocardiogram analysis; J.W.E., J.H.v.B, T.S.L., S.R.H., J.P.L., A.C.C. and A.A.L. assisted with data interpretation and edited the manuscript.
Corresponding author
Additional information
Reviewer Information: Nature thanks M. Murphy and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 shRNA knockdown of NCLX and biophysical characterization in non-excitable cells.
a, qPCR analysis of SLC8B1 mRNA expression in shRNA stable knockdown HeLa cell lines versus scramble shRNA control cell line (scr. con). b, mCa2+ transients recorded in cells expressing the genetic mCa2+ sensor, mitycam, during histamine treatment (100 μM). Mean intensity shown as solid lines, thin dashed lines display s.e.m. c, mCa2+ peak amplitude. d, Per cent mCa2+ efflux versus scramble shRNA control. n = 42–49 cells per group. e–g, HeLa cells were loaded with the Ca2+ sensor, Fura-FF, and the Δψ sensor, JC-1, permeabilized with digitonin (dg) and treated with SERCA inhibitor, thapsigargin (thaps) for simultaneous ratiometric monitoring during repetitive additions of 5 μM Ca2+ (black arrows). FCCP was used to collapse Δψ at the conclusion of each experiment. h, mCa2+ uptake capacity versus scramble shRNA control cells following 20 μM Ca2+ (fourth pulse). i, JC-1-derived Δψ before FCCP addition. n = 3 experiments per assay; *P < 0.05, ***P < 0.001 versus scramble shRNA control.
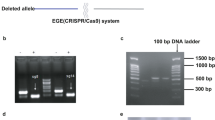
Extended Data Figure 2 Characterization of Slc8b1 conditional knockout following tamoxifen administration.
a, Images of chimeric founder mice and estimated per cent chimerism, black coat colour correlates with mutant ES cell contribution to development. b, Image of gel with PCR genotyping results for Slc8b1 loxP targeted mice. c, Left ventricular end-systolic dimension (LVESD) three days after tamoxifen administration. d, Curve fitting of mitochondrial swelling traces after addition of 500 μM Ca2+. e, Representative images of MitoSOX Red-stained ACMs (scale bar, 40 μm). f, Fold change in MitoSOX Red intensity three days after tamoxifen administration. n = 42–52 ACMs per group. g, Representative ECG recordings of Slc8b1fl/fl × MCM mice (n = 4–7) at baseline, three days and five days after tamoxifen treatment, and day of sinus arrest. h, Quantitation of PR interval. i, Quantitation of QRS interval. j, Quantitation of heart rate (HR). BPM, beats per minute. k, Western blot of proposed MPTP components from heart protein lysate: CypD and ANT, VDAC shown in Fig. 1d. l, Size distribution (perimeter) of mitochondria analysed in electron microscopy images three days after tamoxifen (minimum 200 mitochondria quantified per mouse, n = 3–4 mice per group). m, Quantification of NAD+/NADH ratio three days after tamoxifen administration (fold change versus control). n = 6–7 per group. n, Western blots of total pyruvate dehydrogenase (PDH E1α) and phosphorylated-PDH (p-PDH) from heart protein lysate. o, Fold change in the ratio of p-PDH/total PDH E1α versus control. n = 3 per group; *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant.
Extended Data Figure 3 Examination of Ca2+ flux following αMHC-Cre-mediated deletion of Slc8b1.
a, Adult heart protein lysate isolated from: Slc8b1fl/fl × αMHC-Cre mice and controls were examined by western blot for NCLX expression and various other proteins involved in mCa2+ exchange: MCU, MCUb, MICU1, EMRE, LETM1, VDAC; and OXPHOS components: ATP synthase subunit α (CV-Sα), complex III subunit core 2 (CIII-C2), complex IV subunit I (CIV-SI), and complex I subunit NDUFB8 (CI-SIV). n = 3 hearts per group. b, c, Series of mitycam mCa2+ transients recorded during pacing (0.1 Hz). d, mCa2+ time to peak amplitude. e, Per cent mCa2+ efflux 10 s after pacing. f, Tetramethylrhodamine, ethyl ester (TMRE) (Δψ) in intact ACM. n = 23–24 ACMs per group. g, h, Representative recordings of iCa2+ transients (Fluo-4) in ACMs paced at 1 Hz with or without isoproterenol (Iso, 100 nM). i, iCa2+ peak amplitude. j, iCa2+ time to peak amplitude. k, iCa2+ time to 50% decay. l, Time to 90% decay. n = 10 cells per group. m, n, Permeabilized ACM mCa2+ uptake rate (m) and per cent uptake (n) after Ca2+ addition. *P < 0.05, **P < 0.01, ***P < 0.001.
Extended Data Figure 4 Characterization of NCLX transgenic mice.
a, b, Series of mitycam mCa2+ transients recorded during pacing (0.1 Hz). c, Tetramethylrhodamine, ethyl ester (TMRE) (Δψ) in intact ACMs. n = 23–36 ACMs per group. d, e, Representative traces of iCa2+ transients (Fluo-4) in ACMs paced at 1 Hz with or without isoproterenol (100 nM). f, iCa2+ peak amplitude. g, iCa2+ time to peak amplitude. h, Time to 50% decay. i, Time to 90% decay (n = 15 cells per group). j, Recording of transients in Digitonin (dig)-permeabilized ACMs loaded with Fura-FF, treated with thapsigargin (thaps) and placed in 20 μM bath Ca2+ before treatment with the MCU inhibitor, Ru360, to quantify the rate of mCa2+ efflux independent of uptake. k–q, Seahorse analysis of mitochondrial oxygen consumption rates (OCR) in ACMs in the presence of pyruvate or palmitate. m, Basal OCR. n, ATP-linked respiration after addition of ATP synthase inhibitor, oligomycin. o, Maximal respiration after addition of protonophore, FCCP. p, Spare respiratory capacity (max − basal). q, Proton leak (post-oligomycin OCR – non-mitochondrial OCR). n = 11–15 per condition. r, Quantification of NAD+/NADH ratio three days after tamoxifen (fold change versus control). n = 4–5 per group. s, Western blot of total pyruvate dehydrogenase (PDH E1α) and phosphorylated-PDH (p-PDH) from heart protein lysate. *P < 0.05, ***P < 0.001.
Extended Data Figure 5 Analysis of NCLX-Tg mice 24 h after ichaemia reperfusion and 4 weeks after myocardial infarction.
a, Quantification of TUNEL+ interstitial cells after ischaemia reperfusion. b–e, B-mode speckle tracking analysis of left ventricular function 4 weeks after myocardial infarction: longitudinal strain (b), longitudinal strain rate (c), radial strain (d), radial strain rate (e). n = 20 tTA and n = 18 NCLX-Tg. f, Lung oedema (wet − dry lung weight). n = 7 per sham group, n = 17 per myocardial infarction group. g, Kaplan–Meier survival curves post myocardial infarction. n = 28 tTA and n = 27 NCLX-Tg. h–l, qPCR quantification of mRNA expression in hearts from sham-treated mice or 4 weeks after myocardial infarction. Nppa, atrial natriuretic peptide; Nppb, brain natriuretic peptide; Spp1, osteopontin; Postn1, periostin; Acta2, smooth muscle α-actin (sham, n = 3 group; myocardial infarction, n = 7 per group). m, Representative images of H&E-stained heart sections from the infarct border zone 4 weeks post myocardial infarction. Scale bar, 400 μm. n, o, qPCR analysis of mRNA expression for inflammatory cytokines 4 weeks post myocardial infarction. Il1b, interleukin-1β; Il6, interleukin-6. n = 3 per sham group, n = 7 per myocardial infarction group. p, Western blot of pyruvate dehydrogenase subunits and phosphorylated-PDH (p-PDH) from heart protein lysate 4 weeks post myocardial infarction. q, Fold-change in the ratio of p-PDH/total PDH E1α versus tTA control. n = 4 per group; *P < 0.05, **P < 0.01, ***P < 0.001, versus αMHC tTA post injury; #P < 0.05, ##P < 0.01 versus sham control.
Supplementary information
Supplementary Figure
This file contains Supplementary Figure 1, the uncropped blots and band density results. (PDF 1699 kb)
Rights and permissions
About this article

Cite this article
Luongo, T., Lambert, J., Gross, P. et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545, 93–97 (2017). https://doi.org/10.1038/nature22082
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature22082
This article is cited by
-
IF1 ablation prevents ATP synthase oligomerization, enhances mitochondrial ATP turnover and promotes an adenosine-mediated pro-inflammatory phenotype
Cell Death & Disease (2023)
-
Identity, structure, and function of the mitochondrial permeability transition pore: controversies, consensus, recent advances, and future directions
Cell Death & Differentiation (2023)
-
Quantification of Cardiomyocyte Contraction In Vitro and Drug Screening by MyocytoBeats
Journal of Cardiovascular Translational Research (2023)
-
Pathological Impact of Tau Proteolytical Process on Neuronal and Mitochondrial Function: a Crucial Role in Alzheimer’s Disease
Molecular Neurobiology (2023)
-
Innovative treatments for congenital heart defects
World Journal of Pediatrics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
