
Abstract
Viruses are the most abundant biological entities on Earth, but challenges in detecting, isolating, and classifying unknown viruses have prevented exhaustive surveys of the global virome. Here we analysed over 5 Tb of metagenomic sequence data from 3,042 geographically diverse samples to assess the global distribution, phylogenetic diversity, and host specificity of viruses. We discovered over 125,000 partial DNA viral genomes, including the largest phage yet identified, and increased the number of known viral genes by 16-fold. Half of the predicted partial viral genomes were clustered into genetically distinct groups, most of which included genes unrelated to those in known viruses. Using CRISPR spacers and transfer RNA matches to link viral groups to microbial host(s), we doubled the number of microbial phyla known to be infected by viruses, and identified viruses that can infect organisms from different phyla. Analysis of viral distribution across diverse ecosystems revealed strong habitat-type specificity for the vast majority of viruses, but also identified some cosmopolitan groups. Our results highlight an extensive global viral diversity and provide detailed insight into viral habitat distribution and host–virus interactions.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


References
Suttle, C. A. Marine viruses—major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812 (2007)
Reyes, A. et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466, 334–338 (2010)
Brum, J. R. et al. Ocean plankton. Patterns and ecological drivers of ocean viral communities. Science 348, 1261498 (2015)
Whitman, W. B., Coleman, D. C. & Wiebe, W. J. Prokaryotes: the unseen majority. Proc. Natl Acad. Sci. USA 95, 6578–6583 (1998)
Reddy, T. B. et al. The Genomes OnLine Database (GOLD) v.5: a metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res. 43, D1099–D1106 (2015)
Chow, C. E. & Suttle, C. A. Biogeography of viruses in the sea. Annu Rev Virol 2, 41–66 (2015)
Rohwer, F. & Edwards, R. The Phage Proteomic Tree: a genome-based taxonomy for phage. J. Bacteriol. 184, 4529–4535 (2002)
Fuhrman, J. A. Marine viruses and their biogeochemical and ecological effects. Nature 399, 541–548 (1999)
Brum, J. R. & Sullivan, M. B. Rising to the challenge: accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 13, 147–159 (2015)
Edwards, R. A., McNair, K., Faust, K., Raes, J. & Dutilh, B. E. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev. 40, 258–272 (2016)
Markowitz, V. M. et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 42, D568–D573 (2014)
Edwards, R. A. & Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 3, 504–510 (2005)
Ivanova, N. et al. A call for standardized classification of metagenome projects. Environ. Microbiol. 12, 1803–1805 (2010)
Hurwitz, B. L. U’Ren, J. M. & Youens-Clark, K. Computational prospecting the great viral unknown. FEMS Microbiol. Lett. (2016)
Ignacio-Espinoza, J. C., Solonenko, S. A. & Sullivan, M. B. The global virome: not as big as we thought? Curr. Opin. Virol. 3, 566–571 (2013)
Lu, H. et al. Membrane biofouling in a wastewater nitrification reactor: Microbial succession from autotrophic colonization to heterotrophic domination. Water Res. 88, 337–345 (2016)
Serwer, P., Hayes, S. J., Thomas, J. A. & Hardies, S. C. Propagating the missing bacteriophages: a large bacteriophage in a new class. Virol. J. 4, 21 (2007)
Varghese, N. J. et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–6771 (2015)
Simmonds, P. Methods for virus classification and the challenge of incorporating metagenomic sequence data. J. Gen. Virol. 96, 1193–1206 (2015)
Hurwitz, B. L., Brum, J. R. & Sullivan, M. B. Depth-stratified functional and taxonomic niche specialization in the ‘core’ and ‘flexible’ Pacific Ocean Virome. ISME J. 9, 472–484 (2015)
Roux, S., Hallam, S. J., Woyke, T. & Sullivan, M. B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife 4, (2015)
Mojica, F. J., Díez-Villaseñor, C., García-Martínez, J. & Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740 (2009)
Andersson, A. F. & Banfield, J. F. Virus population dynamics and acquired virus resistance in natural microbial communities. Science 320, 1047–1050 (2008)
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007)
Lum, A. G. et al. Global transcription of CRISPR loci in the human oral cavity. BMC Genomics 16, 401 (2015)
Bailly-Bechet, M., Vergassola, M. & Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 17, 1486–1495 (2007)
Goren, M. G., Yosef, I. & Qimron, U. Programming Bacteriophages by Swapping Their Specificity Determinants. Trends Microbiol. 23, 744–746 (2015)
Salmond, G. P. & Fineran, P. C. A century of the phage: past, present and future. Nat. Rev. Microbiol. 13, 777–786 (2015)
Holmfeldt, K., Middelboe, M., Nybroe, O. & Riemann, L. Large variabilities in host strain susceptibility and phage host range govern interactions between lytic marine phages and their Flavobacterium hosts. Appl. Environ. Microbiol. 73, 6730–6739 (2007)
Peters, D. L., Lynch, K. H., Stothard, P. & Dennis, J. J. The isolation and characterization of two Stenotrophomonas maltophilia bacteriophages capable of cross-taxonomic order infectivity. BMC Genomics 16, 664 (2015)
Emerson, J. B. et al. Virus-host and CRISPR dynamics in archaea-dominated hypersaline Lake Tyrrell, Victoria, Australia. Archaea 2013, 370871 (2013)
Tschitschko, B. et al. Antarctic archaea-virus interactions: metaproteome-led analysis of invasion, evasion and adaptation. ISME J. 9, 2094–2107 (2015)
Breitbart, M. & Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 13, 278–284 (2005)
Dinsdale, E. A. et al. Functional metagenomic profiling of nine biomes. Nature 452, 629–632 (2008)
Breitbart, M., Miyake, J. H. & Rohwer, F. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol. Lett. 236, 249–256 (2004)
Salazar, G. et al. Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J. 10, 596–608 (2016)
Abeles, S. R. & Pride, D. T. Molecular bases and role of viruses in the human microbiome. J. Mol. Biol. 426, 3892–3906 (2014)
Wylie, K. M. et al. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol. 12, 71 (2014)
Robles-Sikisaka, R. et al. Association between living environment and human oral viral ecology. ISME J. 7, 1710–1724 (2013)
Mukherjee, S., Huntemann, M., Ivanova, N., Kyrpides, N. C. & Pati, A. Large-scale contamination of microbial isolate genomes by Illumina PhiX control. Stand. Genomic Sci. 10, 18 (2015)
Bondy-Denomy, J. & Davidson, A. R. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J. Microbiol. 52, 235–242 (2014)
Short, C. M. & Suttle, C. A. Nearly identical bacteriophage structural gene sequences are widely distributed in both marine and freshwater environments. Appl. Environ. Microbiol. 71, 480–486 (2005)
Kyrpides, N. C., Eloe-Fadrosh, E. A. & Ivanova, N. N. Microbiome data science: understanding our microbial planet. Trends Microbiol. 24, 425–427 (2016)
Huntemann, M. et al. The standard operating procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP v.4). Stand. Genomic Sci. 10, 86 (2015)
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44 (D1), D279–D285 (2016)
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44 (D1), D457–D462 (2016)
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010)
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002)
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002)
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011)
Dick, G. J. et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10, R85 (2009)
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009)
Huson, D. H. & Scornavacca, C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 61, 1061–1067 (2012)
Merchant, N. et al. The iPlant Collaborative: Cyberinfrastructure for Enabling Data to Discovery for the Life Sciences. PLoS Biol. 14, e1002342 (2016)
Roux, S., Enault, F., Hurwitz, B. L. & Sullivan, M. B. VirSorter: mining viral signal from microbial genomic data. PeerJ 3, e985 (2015)
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009)
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006)
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000)
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002)
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003)
Bland, C. et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8, 209 (2007)
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16 (2004)
Dutilh, B. E. et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 5, 4498 (2014)
Aziz, R. K., Dwivedi, B., Akhter, S., Breitbart, M. & Edwards, R. A. Multidimensional metrics for estimating phage abundance, distribution, gene density, and sequence coverage in metagenomes. Front. Microbiol. 6, 381 (2015)
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012)
Acknowledgements
We thank A. Visel and H. Maughan for critical reading and feedback, A. Pati for help in earlier versions, and the IMG and GOLD teams for their support. This work was conducted by the US Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, under contract number DE-AC02-05CH11231 and used resources of the National Energy Research Scientific Computing Center, supported by the Office of Science of the US Department of Energy.
Author information
Authors and Affiliations
Contributions
D.P.E., N.N.I., and N.C.K. conceived and led the study. All authors participated in the analysis and interpretation of data. D.P.E., E.E.F., E.R., N.N.I., and N.C.K. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks C. A. Suttle and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
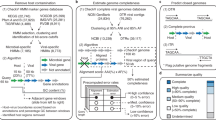
Extended Data Figure 1 Detailed workflow for the identification of viral sequences from metagenomic data.
a, Overview of the acquisition and filtering of viral protein families in two rounds and their use for the identification of metagenomic viral contigs larger than 5 kb. In the first round, proteins from 2,300 double-stranded DNA viruses were grouped into 16,000 protein families, which were aligned to generate Hidden Markov Models (HMMs). These HMMs were used in combination with analysis of k-mer composition and phylogenetic analysis of DNA-dependent RNA polymerase genes to identify 1,843 high-confidence metagenome viral contigs. b, c, These contigs were validated by manual analysis (b) and the proteins from this set were combined with the isolate viral proteins to generate a final set of 25,000 viral protein families (c). d, HMMs generated from alignment of these protein families were used to identify 125,842 metagenomic viral contigs. Processing steps detailed in b–d are described in the Methods. The final mVCs were then grouped and assigned to their hosts via CRISPR–Cas spacer matches and viral tRNA matches against isolate microbes (not shown in this figure).
Extended Data Figure 2 Identification of metagenomic viral contigs via binning and DNA-dependent RNA polymerase alignment.
a–c, Three distinct metagenomic examples of tetranucleotide Emergent Self Organizing Maps (ESOM) as a binning method for identification of candidate viral sequences in metagenome data sets. Tetranucleotide binning of metagenomic samples (full list in Supplementary Table 1) was used to identify highly divergent viral sequences, which were left undetected using viral protein families generated from isolate viruses. Each dot on the maps represents a 10 kb fragment of a metagenomic scaffold longer than 20 kb. ‘Bubbles’ (ESOM structures) correspond to fragments with similar tetranucleotide composition probably originating from the same genome. Red dots represent viral sequences detected by viral protein families generated for isolate viruses; white dots represent highly divergent viral sequences with no hits to viral protein families. a, ESOM of freshwater sample (combined assembly of freshwater microbial communities from Lake Mendota and Trout Bog Lake, IMG identifier 3300000553) shows 2 putative viral sequences previously unidentified (IMG scaffold identifiers 10001161 and 10001271). b, ESOM of marine sample (marine microbial communities from Delaware Coast, sample from Delaware MO Spring March 2010, IMG identifier 3300000116) shows 2 putative viral sequences sequences (IMG scaffold identifiers c10000689 and c10000429). c, ESOM of hydrothermal vent sample (black smokers hydrothermal plume microbial communities from Abe, Lau Basin, Pacific Ocean, IMG identifier 3300001681) showing 2 viral sequences (IMG scaffold identifiers 10000222 and 10000095). Metagenome samples can be found in IMG using IMG identifiers and ‘Quick Search’ or ‘Genome Search’ tools; metagenome scaffolds can be using scaffold identifier and ‘Scaffold Search’ tool on the respective ‘Microbiome Details’ page. d, e, DNA-dependent RNA polymerase genes of likely viral origin from metagenomic sequences longer than 5 kb. d, Hidden Markov Models (HMMs) were built for sequences corresponding to α, β, and β' subunits of bacterial DNA-dependent RNA polymerase for a representative set of 2,551 cellular organisms (archaea, bacteria, and eukaryotes) and viruses. These models were used to search the proteins encoded by metagenomic contigs longer than 5 kb and the proteins with hits were aligned against the HMMs. A total of 7,437 nearly full-length metagenomic sequences were combined with 2,551 reference sequences to reconstruct the phylogenetic tree using FastTree tool. Two distinct branches on this tree were separated from the sequences from cellular organisms and included RNA polymerase genes from eukaryotic viruses (green box) and putative phage sequences with domain structure similar to that of bacterial RNA polymerase (red box, marked with double asterisk). Only 122 out of the 400 contigs in the eukaryotic viral RNA polymerase branch were captured by isolate protein families. e, Detailed view of the RNA polymerase tree branch with putative phage sequences. Metagenome contigs detected as viral by viral protein families and by spacer hits are marked with a square or circle next to it. Gene structure for selected contigs (IMG chromosomal neighbourhood view) is shown in the boxes. In the examples, genes are coloured based on predicted function category (using Clusters of Orthologous Genes prediction) and are specified in the figure. White-coloured genes correspond to those with hypothetical or unknown function.
Extended Data Figure 3 Benchmarking of vHMM-based pipeline and VirSorter on synthetic metagenome data.
Precision (solid lines) and recall (dotted lines) for vHMM pipeline (red) and VirSorter (blue) is plotted against the length of sequence fragments in base pairs. The percentage of contigs detected as viral, but which have at least 10 kb of host sequence is shown by dashed lines for vHMM pipeline (red) and VirSorter (blue).
Extended Data Figure 4 Detailed gene content of singular metagenomic viral contigs examples.
a, Gene content of the metagenomic partial viral genome with the lowest gene coverage by viral protein families. This length of the partial viral genome is 81,542 bp (guanine and cytosine (GC) content of 43%; 163 total genes) and was identified from a bovine rumen metagenome (IMG scaffold identifier, rumenHiSeq_NODE_3763566_len_81492_cov_5_518198; IMG metagenome identifier, 2061766007). White-coloured genes correspond to those with hypothetical or unknown function. Only 3% of the genes were covered by VPFs. b, Gene content of the largest closed viral genome identified to date. The length of the closed (circular) viral genome is 596,617 bp (GC, 40%; 1,148 total genes) and was identified from a bioreactor metagenome (IMG scaffold id: D1draft_1000006, from Bioreactor L1-648F-DHS sludge microbial communities sample). Predicted gene function is coloured based on Clusters of Orthologous Genes. Black triangles indicate tRNAs sequences (a, b). A total of 11% of the genes were covered by VPFs. Specific viral genes distributed across the genome are boxed in red, identified with a number, and described in the legend table. The detailed information of the whole gene content of this viral genome is located in Supplementary Table 11.
Extended Data Figure 5 Viral group clustering method.
a, Parameters used in the clustering of viral sequences. We used all 5,042 reference isolate viral genomes (iVGs) to group them using single-linkage hierarchical clustering (SLC) with different combinations of AAI and AF values to validate the clustering approach. The thresholds for AAI and AF were set at 90% and 50%, respectively, (boxed in purple) and were selected based on the accurate grouping of iVGs that was in agreement at the genus level, and the vast majority at the species level, according to the ICTV classification system (Supplementary Information). Further, these thresholds reduced the number of total connections (green line referred to secondary y axis) compared with lower AAI thresholds, without altering the total number of singletons and viral groups created (red and light green bars referred to primary y axis, respectively), as well as the average number of members per viral group (shown at the bottom of the figure). b, Size distribution of viral groups. Distribution of the 66,696 viral genomes clustered into 18,470 viral groups. Number of viral members (spanning from 2 to 365) per viral groups is shown. c–e, The cytoscape visualization of some viral groups. c, Major reference isolated viral groups created using SLC with AAI and AF values of 90% and 50%, respectively. Cytoscape force-directed (unweighted) layout option was used to visualize these groups. Black nodes represent isolated viral genomes whereas orange and green nodes represent metagenomic viral contigs clustered with isolates from host-associated and environmental samples, respectively. Group edges connect viral groups based on the above cutoffs. d, The four largest viral groups created from metagenomic viral contigs (containing 365, 201, 165, and 152 members, respectively). Specific habitat information of the samples as well as the viral group identifier is shown in the figure. e, Examples of viral groups (vg_2932 and vg_2864) containing proto-spacers (indicated by green circles) found in the CRISPR–Cas system of the indicated bacterial taxon. All the metagenomic viral contigs clustered in both viral groups were found in the same habitat subtype: human oral samples for vg_2932, and human faecal samples for vg_2864 (with a sole exception in the latter group that derived from an oral sample, indicated with a red arrow).
Extended Data Figure 6 Verification of viruses identified with broad-host range.
a, b, Alignments of all contigs found in the IMG database containing any of the 3 spacer matches present in a viral group potentially infecting 2 different phyla or any of the 7 spacer matches present in a viral group potentially infecting 3 different families are shown in a and b, respectively. Alignments were performed by mapping all the matches (48 for a, and 128 for b; named with an IMG scaffold identifier) to a viral representative using the ‘map to reference’ package of Geneious software (http://www.geneious.com). Black lines represent 100% sequence identity to the reference virus. The location of the 3 spacers (that derived from 2 different phyla) in a as well as the 7 spacers (that derived from 3 different families) in b is indicated with triangles with different colours. Spacer sequences, as well as the genomes that contain them in a CRISPR locus is boxed at the bottom.
Extended Data Figure 7 Habitat type specificity of all viral diversity and specific examples.
a, Distribution of the presence of the total viral diversity of metagenomic viral contigs (viral groups and singletons) across distinct number of habitat types. A total of 85.9% of all viral diversity resided in a single habitat type (either as a singleton 19.7%, as a viral group found in a single sample 1.8%, or as a viral group found in 2 or more samples 64.4%), whereas only a small fraction (0.31% of all mVCs) were found in 4 or more different habitat types. b, c, Examples of viral groups found in diverse samples across different oceanic zones and provinces. Presence of a single viral group across distinct marine samples based on average coverage values (red bars; y axis on the left) and total percentage of the viral sequence length recovered per sample (purple line; y axis on the right). Samples were grouped by marine zones and indicate the percentage of the total samples per zone. b, Representative of viral group 2463 (IMG taxon id: 3300001450 and IMG scaffold id: JGI24006J15134_100002847) was found exclusively in marine biomes at depth and with reduced exposure to sunlight (across 95% of all twilight samples and in 44% of deep ocean samples). c, Representative of viral group 10643 (IMG taxon id: 3300000216 and IMG scaffold id: SI53jan11_150mDRAFT_c1002499) detected preferentially across coastal water samples (28% of all samples of this zone, preferentially in oxygen minimum zones), but also present in twilight, deep ocean, and hydrothermal vent samples. This viral group was identified as a SUP05-infecting phage. The genes of the viral contig representatives were coloured by the phylogenetic distribution of the best hit in the database (white, unknown; green, Proteobacteria; blue, Chlorophyta, red, unclassified virus; turquoise, Firmicutes; purple, Deinococcus). d, e, The distribution of viral sequences of distinct body sub-sites across different individuals. Hierarchical clustering (average linkage using Jaccard distance) was used for both axes (samples and individuals) across ‘large intestine’ (d) and ‘oral’ metagenomes (e), respectively (top chart in both panels). Presence or absence of viral groups or singletons per sample is colour-coded as red or blue, respectively. The line chart of both panels show the percentage of viral sharing for >50%, 50–10%, and <10% of the individuals (vertical lines) highlighting in red boxes the percentage of viral sharing for >80% as well as viral sequences only present in a single individual.
Extended Data Figure 8 Alignment of broad-host specificity prophage in 20 isolate genomes in IMG using ‘Gene Neighborhood’ tool.
The gene ‘adenine-specific DNA methyltransferase’ is used as an anchor for the alignment (in red). Genes are coloured according to COG cluster annotation, with light yellow representing genes without COG assignment. Blue boxes highlight likely cargo genes inserted in prophage genomes. These include: (1) alkyl hydroperoxide reductase system in Dehalogenimonas lykanthroporepellens, Desulfococcus biacutus and Geobacter sulfurreducens, (2) efflux ABC transporter in Desulfoarculus baarsii and Desulfobacterium anilini, (3) possible secondary metabolite biosynthesis genes in Desulfovibrio aespoenensis, (4) restriction system in Desulfovibrio paquesii and Geoalkalibacter subterraneous, (5) methionine synthase in Desulfovibrio sp. L21-Syr-AB, (6) molybdate ABC transporter in Desulfomicrobium thermophilum, (7) ABC transporter involved in multi-copper enzyme maturation in Desulfovibrio alkalitolerans; and (8) likely antibiotic resistance cassette in Geobacter soli. Details in Supplementary Table 24.
Extended Data Figure 9 Distribution of hits to broad-host prophage and its potential hosts in metagenomic samples.
The hits to prophage sequences and host marker genes (RNA polymerase subunits and ribosomal proteins) were identified by BLASTn with e-value 1.0 × 1050; 90% nucleotide identity and cumulative alignment length of at least 10% of the length of the prophage or concatenated marker genes. Metagenome samples grouped by habitat are shown on the y axis; boxes correspond to broad environmental categories. Red box surrounds non-human host-associated samples (worm and termite symbionts), green box surrounds environmental samples (aquatic and terrestrial), blue box surrounds engineered samples (wastewater and bioreactors). Average coverage of the prophage and concatenated host marker genes is plotted on the x axis.
Extended Data Figure 10 Global connectivity of viral diversity from different habitat types.
Geographic location of metagenomic samples containing the same viral groups and singletons represented by a white connecting line across metagenomes from different habitats. Only samples sharing 2 or more viral groups or singletons that are more distant than 10 pixels (area shown as a red square in the figure) are connected. The colours of the samples (circles) indicate the habitat type according with the legend. A freely available equirectangular projection of the world map was used as a background image (http://visibleearth.nasa.gov/view.php?id=57752).
Supplementary information
Supplementary Information
This file contains Supplementary Results, Supplementary References and full legends for Supplementary Tables 1-28. (PDF 512 kb)
Supplementary Data
This file contains Supplementary Tables 1-28 – see the Supplementary Information document for full table legends. (XLSX 21221 kb)
Rights and permissions
About this article

Cite this article
Paez-Espino, D., Eloe-Fadrosh, E., Pavlopoulos, G. et al. Uncovering Earth’s virome. Nature 536, 425–430 (2016). https://doi.org/10.1038/nature19094
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature19094
This article is cited by
-
Exploring virus-host-environment interactions in a chemotrophic-based underground estuary
Environmental Microbiome (2024)
-
Biogeographic patterns and drivers of soil viromes
Nature Ecology & Evolution (2024)
-
COBRA improves the completeness and contiguity of viral genomes assembled from metagenomes
Nature Microbiology (2024)
-
Diversity and potential host-interactions of viruses inhabiting deep-sea seamount sediments
Nature Communications (2024)
-
Complete genome sequence of a novel mitovirus isolated from the fungus Fusarium oxysporum f. sp. ginseng causing ginseng root rot
Archives of Virology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
